
CIBINQO tabletti, kalvopäällysteinen 50 mg, 100 mg, 200 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
CIBINQO LÄÄKKEEN MÄÄRÄÄJÄN OPAS
Potilas
Cibinqo (abrositinibi) POTILASKORTTI
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Cibinqo 50 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 50 mg abrositinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 1,37 mg laktoosimonohydraattia.
Cibinqo 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg abrositinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 2,73 mg laktoosimonohydraattia.
Cibinqo 200 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 200 mg abrositinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 5,46 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Cibinqo on tarkoitettu keskivaikean tai vaikean atooppisen ihottuman hoitoon aikuisille ja vähintään 12-vuotiaille nuorille, joille harkitaan systeemistä hoitoa.
Ehto
Hoito tulee toteuttaa indikaation mukaiseen hoitoon perehtyneen lääkärin aloittamana ja valvonnassa. Hoitoa saaville potilaille tulee antaa erityinen potilaskortti.
Annostus ja antotapa
Hoito tulee toteuttaa atooppisen ihottuman diagnosointiin ja hoitoon perehtyneen terveydenhuollon ammattilaisen aloittamana ja valvonnassa.
Annostus
Suositeltu aloitusannos on 100 mg tai 200 mg kerran vuorokaudessa potilaan yksilöllisten ominaisuuksien mukaan:
- Aloitusannosta 100 mg kerran vuorokaudessa suositellaan potilaille, joilla on tavanomaista suurempi laskimotromboembolian, merkittävien sydän- ja verisuonitapahtumien (MACE) ja syövän riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilaan hoitovaste annoksella 100 mg kerran vuorokaudessa ei ole riittävä, annosta voidaan suurentaa 200 mg:aan kerran vuorokaudessa.
- Annos 200 mg kerran vuorokaudessa voi sopia potilaille, joiden laskimotromboembolian, merkittävien sydän- ja verisuonitapahtumien tai syövän, johon liittyy suuri tautitaakka, riski ei ole tavanomaista suurempi tai potilaille, joiden hoitovaste annoksella 100 mg kerran vuorokaudessa ei ole riittävä. Kun hoitotasapaino on saavutettu, annosta on pienennettävä 100 mg:aan kerran vuorokaudessa. Jos hoitotasapaino ei säily annoksen pienentämisen jälkeen, voidaan harkita hoidon jatkamista uudelleen annoksella 200 mg kerran vuorokaudessa. Nuorille (12–17-vuotiaille), jotka painavat 25 – < 59 kg, suositellaan aloitusannosta 100 mg kerran vuorokaudessa. Jos potilaan hoitovaste annoksella 100 mg kerran vuorokaudessa ei ole riittävä, annosta voidaan suurentaa 200 mg:aan kerran vuorokaudessa. Vähintään 59 kg painaville nuorille voi sopia aloitusannos 100 mg tai 200 mg kerran vuorokaudessa.
Ylläpitohoitoa varten on harkittava pienintä tehokasta annosta.
Hoidon keskeyttämistä on harkittava potilailla, joilla ei ilmene näyttöä terapeuttisesta hyödystä 24 viikon aikana.
Cibinqo-valmistetta voidaan käyttää atooppisen ihottuman hoitoon käytettävien paikallislääkitysten kanssa tai ilman niitä.
Seuranta laboratoriokokein
Taulukko 1. Laboratoriomääritykset ja seurantaa koskeva ohje | ||
Laboratoriomääritykset | Seurantaa koskeva ohje | Toimenpide |
Täydellinen verenkuva, mukaan lukien trombosyyttimäärä, absoluuttinen lymfosyyttimäärä (B-Lymf), absoluuttinen neutrofiilimäärä (B-Neut) ja hemoglobiini (Hb). | Ennen hoidon aloittamista, 4 viikkoa aloittamisen jälkeen ja sen jälkeen potilaan rutiinihoidon mukaisesti. | Trombosyytit: Hoito on lopetettava, jos trombosyyttimäärä on < 50 × 109/l. |
B-Lymf: Hoito on keskeytettävä, jos B-Lymf on < 0,5 × 109/l ja se voidaan aloittaa uudelleen, kun B-Lymf palautuu tämän arvon yläpuolelle. Hoito on lopetettava, jos tämä arvo varmistuu. | ||
B-Neut: Hoito on keskeytettävä, jos B-Neut on < 1 × 109/l ja se voidaan aloittaa uudelleen, kun B-Neut palautuu tämän arvon yläpuolelle. | ||
Hb: Hoito on keskeytettävä, jos Hb on < 80 g/l ja se voidaan aloittaa uudelleen, kun Hb palautuu tämän arvon yläpuolelle. | ||
Lipidiparametrit | Ennen hoidon aloittamista, 4 viikkoa aloittamisen jälkeen ja sen jälkeen huomioiden potilaan sydän- ja verisuonitautien riskit ja hyperlipidemiaa koskevat kliiniset suositukset. | Potilaita on seurattava hyperlipidemiaa koskevien kliinisten suositusten mukaisesti. |
Hoidon aloittaminen
Hoitoa ei pidä aloittaa potilaille, joiden trombosyyttimäärä on < 150 × 109/l, absoluuttinen lymfosyyttimäärä (B-Lymf) on < 0,5 × 109/l, absoluuttinen neutrofiilimäärä (B-Neut) on < 1,2 × 109/l tai joiden hemoglobiiniarvo on < 100 g/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostelun keskeyttäminen
Jos potilaalle kehittyy vakava infektio, sepsis tai opportunistinen infektio, annostelun keskeyttämistä on harkittava, kunnes infektio on saatu hallintaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostelun keskeyttäminen voi olla tarpeen taulukossa 1 kuvattujen poikkeavien laboratorioarvojen hoitamiseksi.
Väliin jääneet annokset
Jos annos jää väliin, potilaita on kehotettava ottamaan annos mahdollisimman pian, paitsi jos seuraavaan annokseen on alle 12 tuntia, jolloin potilaan ei pidä ottaa väliin jäänyttä annosta. Tämän jälkeen annostelua on jatkettava tavanomaisen aikataulun mukaisesti.
Yhteisvaikutukset
Potilaille, jotka saavat sekä sytokromi P450 (CYP) 2C19:ää voimakkaasti että CYP2C9:ää kohtalaisesti estäviä lääkevalmisteita tai yksistään CYP2C19:n voimakkaita estäjiä (esim. fluvoksamiini, flukonatsoli, fluoksetiini ja tiklopidiini), suositeltu annos on puolitettava 100 mg:aan tai 50 mg:aan kerran vuorokaudessa (ks. kohta Yhteisvaikutukset).
Hoitoa ei suositella samanaikaisesti käytettävien kohtalaisen voimakkaiden tai voimakkaiden CYP2C19/CYP2C9-entsyymien induktoreiden kanssa (esim. rifampisiini, apalutamidi, efavirentsi, entsalutamidi, fenytoiini) (ks. kohta Yhteisvaikutukset).
Potilaille, jotka saavat haponestolääkkeitä (esim. antasidit, protonipumpun estäjät ja H2-reseptorin salpaajat), abrositinibiannosta 200 mg kerran vuorokaudessa tulee harkita (ks. kohta Yhteisvaikutukset).
Erityiset potilasryhmät
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä munuaisten vajaatoiminta, eli glomerulusten laskennallinen suodatusnopeus (eGFR) on 60 – < 90 ml/min.
Potilaille, joilla on keskivaikea (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta, abrositinibin suositeltu annos on puolitettava, annokseen 100 mg tai 50 mg kerran vuorokaudessa (ks. kohta Farmakokinetiikka).
Potilaille, joilla on vaikea (eGFR < 30 ml/min) munuaisten vajaatoiminta, suositeltu aloitusannos on 50 mg kerran vuorokaudessa. Suurin vuorokausiannos on 100 mg (ks. kohta Farmakokinetiikka).
Abrositinibia ei ole tutkittu potilailla, joilla on loppuvaiheen munuaissairaus (ESRD) ja jotka saavat munuaiskorvaushoitoa.
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä (Child Pugh A) tai keskivaikea (Child Pugh B) maksan vajaatoiminta. Abrositinibi on vasta-aiheista potilaille, joilla on vaikea (Child Pugh C) maksan vajaatoiminta (ks. kohta Vasta-aiheet).
Iäkkäät
Suositeltu annos ≥ 65-vuotiaille potilaille on 100 mg kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Cibinqo-valmisteen turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste otetaan suun kautta kerran päivässä ruoan kanssa tai tyhjään mahaan suunnilleen samaan aikaan joka päivä.
Potilailla, joilla ilmenee pahoinvointia, tablettien ottaminen ruoan kanssa voi lievittää pahoinvointia.
Tabletit on nieltävä kokonaisina veden kanssa. Niitä ei pidä jakaa, murskata tai pureskella, koska näitä antotapoja ei ole tutkittu kliinisissä tutkimuksissa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Aktiiviset vakavat systeemiset infektiot, kuten tuberkuloosi (TB) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
- Raskaus ja imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Abrositinibia tulee käyttää seuraaville potilasryhmille vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä:
|
Infektiot/vakavat infektiot
Vakavia infektioita on raportoitu abrositinibia saavilla potilailla. Kliinisissä tutkimuksissa yleisimmin ilmenneitä vakavia infektioita olivat Herpes simplex, Herpes zoster ja keuhkokuume (ks. kohta Haittavaikutukset).
Iäkkäillä ja diabetesta sairastavilla infektioiden ilmaantuvuus on yleensä tavanomaista suurempaa, minkä vuoksi iäkkäitä ja diabetespotilaita hoidettaessa on noudatettava varovaisuutta. Abrositinibia tulisi käyttää 65-vuotiaille ja sitä vanhemmille potilaille vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä (ks. kohta Annostus ja antotapa).
Hoitoa ei saa aloittaa potilaille, joilla on aktiivinen, vakava systeeminen infektio (ks. kohta Vasta-aiheet).
Hoidon riskejä ja hyötyjä on harkittava ennen abrositinibihoidon aloittamista potilaille:
- joilla on krooninen tai toistuva infektio
- jotka ovat altistuneet tuberkuloosille
- joilla on ollut vakava tai opportunistinen infektio
- jotka ovat asuneet tai matkustaneet alueilla, joilla tuberkuloosi tai mykoosit ovat endeemisiä; tai
- joilla on taustasairauksia, jotka voivat altistaa heidät infektiolle.
Potilaita pitää seurata huolellisesti infektion merkkien ja oireiden kehittymisen varalta abrositinibihoidon aikana ja sen jälkeen. Jos potilaalle kehittyy uusi infektio hoidon aikana, hänelle on tehtävä pikaisesti täydelliset diagnostiset testit ja asianmukainen antimikrobihoito on aloitettava. Potilasta on seurattava tarkasti ja hoito on keskeytettävä tilapäisesti, jos potilas ei reagoi tavanomaiseen hoitoon.
Tuberkuloosi
Kliinisissä abrositinibitutkimuksissa havaittiin tuberkuloosia. Potilaat on testattava tuberkuloosin toteamiseksi ennen hoidon aloittamista ja on harkittava vuosittaista testausta potilaille alueilla, joissa tuberkuloosi on hyvin endeeminen. Abrositinibia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Jos potilaalla on hiljattain todettu piilevä tuberkuloosi tai aikaisemmin hoitamaton piilevä tuberkuloosi, piilevän tuberkuloosin ehkäisyhoito on aloitettava ennen hoidon aloittamista.
Virusten uudelleenaktivoituminen
Virusten uudelleenaktivoitumista, mukaan lukien herpesviruksen uudelleenaktivoitumista (esim. Herpes zoster, Herpes simplex) raportoitiin kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Vyöruusu (Herpes zoster) ‑infektioiden määrä oli suurempi 200 mg:n annoksia saaneilla potilailla, vähintään 65- vuotiailla potilailla sekä potilailla, joilla oli anamneesissa vyöruusu (Herpes zoster), ennen tapahtumaa varmistettu B-Lymf < 1 × 109/l ja lähtötilanteessa vaikea atooppinen ihottuma (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy vyöruusu (Herpes zoster), hoidon tilapäistä keskeyttämistä on harkittava, kunnes vyöruusu häviää.
Virushepatiitin seulonta on tehtävä kliinisten suositusten mukaisesti ennen hoidon aloittamista ja hoidon aikana. Potilaat, joilla oli näyttöä aktiivisesta B-hepatiitti- tai C-hepatiitti-infektiosta (positiivinen C-hepatiitti-PCR), suljettiin pois kliinisistä lääketutkimuksista (ks. kohta Farmakokinetiikka). Potilaille, jotka olivat negatiivisia B-hepatiitti-pinta-antigeenin suhteen, positiivisia B-hepatiitti-ydinantigeenin suhteen ja positiivisia B-hepatiitti-pinta-vasta-aineen suhteen, tehtiin B-hepatiittivirus (HBV) -DNA-testi. Potilaat, joiden HBV-DNA-arvo oli alemman määritysrajan (LLQ) yläpuolella, suljettiin pois tutkimuksesta. Potilaille, joiden HBV-DNA-arvo oli negatiivinen tai alle LLQ:n, aloitettiin hoito ja potilaiden HBV-DNA-arvoa seurattiin. Jos HBV-DNA havaitaan, on neuvoteltava maksasairauksiin erikoistuneen lääkärin kanssa.
Rokotus
Abrositinibihoitoa saavien potilaiden rokotusvasteesta ei ole saatavilla tietoja. Eläviä, heikennettyjä taudinaiheuttajia sisältävien rokotteiden käyttöä on vältettävä hoidon aikana tai juuri sitä ennen. Ennen tällä lääkevalmisteella annettavan hoidon aloittamista suositellaan, että potilaiden rokotukset tuodaan ajan tasalle, myös ennaltaehkäisevät Herpes zoster -rokotukset, nykyisten rokotussuositusten mukaisesti.
Laskimotromboembolia
Abrositinibia käyttävillä potilailla on raportoitu syviä laskimotukoksia (SLT) ja keuhkoembolioita (KE) (ks. kohta Haittavaikutukset).
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibia saaneilla potilailla laskimotromboembolian, mukaan lukien syvä laskimotukos ja keuhkoembolia, ilmaantuvuus oli suurempi ja annosriippuvainen.
Käytettäessä 200 mg:n abrositinibiannosta laskimotromboembolian ilmaantuvuus oli suurempi kuin 100 mg:n abrositinibiannoksella.
Potilaille, joilla on sydän- ja verisuonitapahtumiin tai syöpään liittyviä riskitekijöitä (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet ”Merkittävät sydän- ja verisuonitapahtumat” ja ”Syöpä”), abrositinibia tulee käyttää vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä.
Abrositinibin käytössä on noudatettava varovaisuutta, jos potilaalla on muita kuin sydän- ja verisuonitapahtumiin tai syöpään liittyviä laskimotromboembolian riskitekijöitä. Muita kuin sydän- ja verisuonitapahtumiin tai syöpään liittyviä laskimotromboembolian riskitekijöitä ovat aiempi laskimotromboembolia, potilaalle tehtävä suuri leikkaus, immobilisaatio, hormonaalisten yhdistelmäehkäisyvalmisteiden tai hormonikorvausvalmisteiden käyttö ja periytyvä hyytymishäiriö.
Potilaat on tutkittava säännöllisesti abrositinibihoidon aikana laskimotromboembolian riskin muutosten arvioimiseksi.
Potilas, jolla on laskimotromboembolian merkkejä ja oireita, on viipymättä tutkittava, ja epäiltäessä potilaalla laskimotromboemboliaa abrositinibihoito on lopetettava sen annostuksesta riippumatta.
Merkittävät sydän- ja verisuonitapahtumat (MACE)
Abrositinibia käyttävillä potilailla on todettu merkittäviä sydän- ja verisuonitapahtumia.
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibia saaneilla potilailla merkittävien sydän- ja verisuonitapahtumien, määriteltynä sydän- ja verisuonitapahtumiin liittyviin kuolemantapauksiin, kuolemaan johtamattomiin sydäninfarkteihin ja kuolemaan johtamattomiin aivohalvauksiin, ilmaantuvuus oli suurempi.
Tästä syystä 65-vuotiaille ja sitä vanhemmille potilaille, pitkään tupakoineille tai aiemmin pitkään tupakoineille potilaille sekä potilaille, joilla on aiemmin ollut ateroskleroottinen valtimotauti tai joilla on muita sydän- ja verisuonitapahtumien riskitekijöitä, abrositinibia tulisi käyttää vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä.
Syöpä (ei-melanoottista ihosyöpää lukuun ottamatta)
JAK-estäjillä, mukaan lukien abrositinibilla, hoidetuilla potilailla on todettu lymfoomia ja muita syöpiä.
Laajassa, satunnaistetussa, aktiivikontrolloidussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) oli mukana vähintään 50-vuotiaita nivelreumapotilaita, joilla oli vähintään yksi sydän- ja verisuonitapahtumien lisäriskitekijä. Siinä havaittiin, että TNF:n estäjiin verrattuna tofasitinibia saaneilla potilailla syöpien, erityisesti keuhkosyövän, lymfooman ja ei-melanoottisen ihosyövän, ilmaantuvuus oli suurempi.
Käytettäessä 200 mg:n abrositinibiannosta syöpien (ei melanoottista ihosyöpää lukuun ottamatta) ilmaantuvuus oli suurempi kuin 100 mg:n abrositinibiannoksella.
65-vuotiaille ja sitä vanhemmille potilaille, pitkään tupakoineille tai aiemmin pitkään tupakoineille sekä potilaille, joilla on muita syöpään liittyviä riskitekijöitä (esim. aktiivinen tai aiemmin sairastettu syöpä), abrositinibia tulee käyttää vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä.
Ei-melanoottinen ihosyöpä
Abrositinibilla hoidetuilla potilailla on todettu ei-melanoottista ihosyöpää. Säännöllistä ihon tutkimista suositellaan kaikille potilaille, erityisesti niille, joilla on tavanomaista suurempi ihosyöpäriski.
Hematologiset poikkeavuudet
Vahvistettu absoluuttinen lymfosyyttimäärä < 0,5 × 109/l ja trombosyyttimäärä < 50 × 109/l havaittiin alle 0,5 %:lla potilaista kliinisissä lääketutkimuksissa (ks. kohta Haittavaikutukset). Abrositinibihoitoa ei pidä aloittaa potilaille, joiden trombosyyttimäärä on < 150 × 109/l, B-Lymf < 0,5 × 109/l, B-Neut < 1,2 × 109/l tai joiden hemoglobiiniarvo on < 100 g/l (ks. kohta Annostus ja antotapa). Täydellinen verenkuva on määritettävä 4 viikon kuluttua hoidon aloittamisesta ja tämän jälkeen potilaan rutiinihoidon mukaisesti (ks. taulukko 1).
Lipidit
Veren lipidiarvojen annosriippuvaista suurenemista raportoitiin abrositinibilla hoidetuilla potilailla verrattuna lumelääkkeellä hoidettuihin potilaisiin (ks. kohta Haittavaikutukset). Lipidiarvot on arvioitava noin 4 viikkoa hoidon aloittamisen jälkeen ja tämän jälkeen potilaan sydän- ja verisuonisairauksien riskin mukaisesti (ks. taulukko 1). Näiden lipidiarvojen suurenemisten vaikutusta sydän- ja verisuonitaudista johtuvaan sairastuvuuteen ja kuolleisuuteen ei ole määritetty. Potilaita, joilla on poikkeavat lipidiarvot, on edelleen seurattava ja hoidettava kliinisten suositusten mukaisesti, johtuen hyperlipidemiaan liittyvistä tunnetuista sydän- ja verisuonisairauksien riskeistä.
Iäkkäät
Iäkkäillä potilailla havaittu turvallisuusprofiili oli samanlainen kuin aikuispopulaatiossa, seuraavin poikkeuksin: suurempi osuus vähintään 65-vuotiaista potilaista keskeytti osallistumisensa kliinisiin lääketutkimuksiin ja heillä oli todennäköisemmin vakavia haittavaikutuksia nuorempiin potilaisiin verrattuna; vähintään 65-vuotiaat potilaat saivat todennäköisemmin alhaisia trombosyytti- ja lymfosyyttiarvoja; Herpes zoster -ilmaantuvuus oli korkeampi vähintään 65-vuotiailla potilailla kuin nuoremmilla potilailla (ks. kohta Haittavaikutukset). Yli 75-vuotiaista potilaista on vain vähän tietoja.
Käyttö 65-vuotiaille ja sitä vanhemmille potilaille
Laajassa, satunnaistetussa tutkimuksessa tofasitinibilla (eräs JAK-estäjä) havaittiin, että tofasitinibin käyttöön 65‑vuotiaille ja sitä vanhemmille potilaille liittyy tavanomaista suurempi merkittävien sydän- ja verisuonitapahtumien, syöpien, vakavien infektioiden ja kuolleisuuden (kaikki syyt) riski. Sen vuoksi abrositinibihoitoa tulisi käyttää näille potilaille vain, jos soveltuvia hoitovaihtoehtoja ei ole käytettävissä.
Immuunivastetta heikentävät sairaudet tai lääkevalmisteet
Potilaat, joilla oli immuunipuutossairauksia tai ensimmäisen asteen sukulainen, jolla oli perinnöllinen immuunipuutostila, suljettiin pois kliinisistä lääketutkimuksista eikä näistä potilaista ole tietoja.
Yhdistelmää biologisten immuunivasteen muuntajien, voimakkaiden immuunisalpaajien, kuten siklosporiini, tai muiden Janus-kinaasin (JAK) estäjien, kanssa ei ole tutkittu. Niiden samanaikaista käyttöä abrositinibin kanssa ei suositella, koska additiivisen immuunisalpauksen riskiä ei voida sulkea pois.
Apuaineet
Laktoosimonohydraatti
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti, eli sen voidaan sanoa olevan "natriumiton".
Yhteisvaikutukset
Muiden lääkevalmisteiden mahdollinen vaikutus abrositinibin farmakokinetiikkaan
Abrositinibi metaboloituu pääasiallisesti CYP2C19- ja CYP2C9-entsyymien välityksellä, ja vähäisemmässä määrin CYP3A4- ja CYP2B6-entsyymien välityksellä, ja sen aktiiviset metaboliitit erittyvät munuaisten kautta ja ovat orgaanisten anionien kuljetusproteiini 3:n (OAT3) substraatteja. Näin ollen abrositinibille ja/tai sen aktiivisille metaboliiteille altistumiseen voivat vaikuttaa lääkevalmisteet, jotka estävät tai indusoivat näitä entsyymejä ja kuljetusproteiinia. Annoksen muuttaminen asianmukaisesti selitetään pääpiirteissään kohdassa Annostus ja antotapa.
Samanaikainen anto CYP2C19/CYP2C9:n estäjien kanssa
Kun 100 mg abrositinibia annettiin samanaikaisesti fluvoksamiinin (voimakas CYP2C19:n ja kohtalainen CYP3A:n estäjä) tai flukonatsolin (voimakas CYP2C19:n, kohtalainen CYP2C9:n ja CYP3A:n estäjä) kanssa, altistuminen abrositinibin aktiiviselle rakenneosalle (ks. kohta Farmakokinetiikka) lisääntyi 91 %:lla fluvoksamiinin kanssa ja 155 %:lla flukonatsolin kanssa, verrattuna antoon yksinään (ks. kohta Annostus ja antotapa).
Samanaikainen anto CYP2C19/CYP2C9:n induktorien kanssa
200 mg:n abrositinibiannoksen anto useiden rifampisiiniannosten kanssa (rifampisiini on CYP‑entsyymien voimakas induktori) sai aikaan sen, että altistuminen abrositinibin aktiiviselle rakenneosalle pieneni noin 56 %:lla (ks. kohta Annostus ja antotapa).
Samanaikainen anto OAT3:n estäjien kanssa
Kun 200 mg abrositinibia annettiin samanaikaisesti probenesidin kanssa (probenesidi on OAT3:n estäjä), altistumiset abrositinibin aktiiviselle rakenneosalle lisääntyivät noin 66 %:lla. Tämä ei ole kliinisesti merkittävää eikä annosta tarvitse muuttaa.
Samanaikainen anto mahan pH-arvoa nostavien valmisteiden kanssa
Annettaessa 200 mg:n abrositinibiannoksen kanssa samanaikaisesti 40 mg famotidiinia, joka on H2‑reseptorin antagonisti, abrositinibin aktiiviselle rakenneosalle altistuminen väheni noin 35 %. Mahan pH-arvoa nostavien antasidien tai protonipumpun estäjien (omepratsoli) vaikutusta abrositinibin farmakokinetiikkaan ei ole tutkittu ja se voi olla samankaltainen kuin famotidiinin käytön yhteydessä on havaittu. Suurempaa 200 mg:n vuorokausiannosta tulee harkita potilaille, joita hoidetaan samanaikaisesti mahan pH-arvoa nostavilla valmisteilla, koska nämä saattavat vähentää abrositinibin tehoa.
Abrositinibin mahdollinen vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Mitään abrositinibin kliinisesti merkittäviä vaikutuksia ei havaittu yhteisvaikutusta koskevissa tutkimuksissa, joissa tutkittiin ehkäisytabletteja (esim. etinyyliestradioli/levonorgestreeli).
Abrositinibi on P-glykoproteiinin (P-gp) estäjä in vitro. Dabigatraanieteksilaatin (P-gp:n substraatti) samanaikainen anto yksittäisen 200 mg:n abrositinibiannoksen kanssa lisäsi dabigatraanin AUCinf ‑arvoa noin 53 % ja Cmax-arvoa noin 40 % yksinään antoon verrattuna. Abrositinibin ja dabigatraanin samanaikaisessa käytössä on noudatettava varovaisuutta. Abrositinibin vaikutusta muiden P-gp:n substraattien farmakokinetiikkaan ei ole arvioitu. Varovaisuutta on noudatettava, koska sellaisten P-gp:n substraattien, joilla on kapea terapeuttinen indeksi, kuten digoksiini, määrä voi lisääntyä.
In vitro abrositinibi on CYP2C19-entsyymin estäjä. Annettaessa 200 mg abrositinibia kerran vuorokaudessa ja samanaikaisesti 10 mg omepratsolia kerta-annoksena omepratsolin AUCinf-arvo suureni noin 189 % ja Cmax-arvo noin 134 %, mikä osoittaa, että abrositinibi on kohtalainen CYP2C19-entsyymin estäjä. Käytettäessä abrositinibia samanaikaisesti sellaisten lääkkeiden kanssa, joilla on kapea terapeuttinen indeksi ja jotka metaboloituvat pääasiassa CYP2C19-entsyymin (esim. S‑mefenytoiini ja klopidogreeli) välityksellä, on noudatettava varovaisuutta. Muiden pääasiassa CYP2C19-entsyymin välityksellä metaboloituvien lääkkeiden (esim. sitalopraami, klobatsaami, essitalopraami ja selumetinibi) annoksen säätäminen kyseisen valmisteen valmistetietojen mukaisesti voi olla tarpeen.
Annettaessa 200 mg abrositinibia kerran vuorokaudessa ja samanaikaisesti 100 mg kofeiinia kerta-annoksena kofeiinin AUCinf-arvo suureni 40 %, mutta Cmax-arvo ei muuttunut, mikä viittaa siihen, että abrositinibi on heikko CYP1A2-entsyymin estäjä. Mitään yleistä annosmuutosta ei voida suositella.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, pitää neuvoa käyttämään tehokasta ehkäisymenetelmää hoidon aikana ja yhden kuukauden ajan viimeisen Cibinqo-annoksen jälkeen. Naisten, jotka voivat tulla raskaaksi, raskauden suunnittelua ja ehkäisyä on rohkaistava.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja abrositibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta. Abrositinibin on osoitettu aiheuttavan embryofetaalista letaalisuutta tiineissä rotissa ja kaneissa, luustomuutoksia tiineiden rottien ja kanien sikiöissä, ja vaikuttavan synnytykseen ja peri/postnataaliseen yksilönkehitykseen rotilla (ks. kohta Prekliiniset tiedot turvallisuudesta). Cibinqo on vasta-aiheinen raskauden aikana (ks. kohta Vasta-aiheet).
Imetys
Abrositinibin esiintymisestä ihmisen rintamaidossa, sen vaikutuksista imetettyyn lapseen, tai sen vaikutuksista maidontuotantoon ei ole tietoja. Abrositinibi erittyi imettävien rottien maitoon. Riskiä vastasyntyneille/pikkulapsille ei voida sulkea pois ja Cibinqo on vasta-aiheinen imetyksen aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Rotilla saatujen löydösten perusteella Cibinqo-valmisteen anto suun kautta voi saada aikaan hedelmällisessä iässä olevien naisten hedelmällisyyden tilapäisen alentumisen. Vaikutukset naarasrottien hedelmällisyyteen olivat palautuvia 1 kuukauden kuluttua siitä, kun abrositinibin anto suun kautta keskeytettiin (ks. kohta Prekliiniset tiedot turvallisuudesta)
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Cibinqo-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Useimmin raportoidut haittavaikutukset ovat pahoinvointi (15,1 %), päänsärky (7,9 %), akne (4,8 %), Herpes simplex (4,2 %), kohonnut veren kreatiinikinaasiarvo (3,8 %), oksentelu (3,5 %), heitehuimaus (3,4 %) ja ylävatsakipu (2,2 %). Yleisimmät vakavat haittavaikutukset ovat infektioita (0,3 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Yhteensä 3 848 potilasta hoidettiin abrositinibilla atooppista ihottumaa koskevissa kliinisissä lääketutkimuksissa. Näistä 3 050 potilasta (edustavat 5 166:ta altistumisen potilasvuotta) otettiin mukaan yhdistettyyn turvallisuusanalyysiin. Yhdistetty turvallisuusanalyysi käsitti 1 997 potilasta, jotka saivat jatkuvasti 200 mg:n abrositinibiannoksia ja 1 053 potilasta, jotka saivat jatkuvasti 100 mg:n abrositinibiannoksia. 2 013 potilaalla oli ainakin 48 viikon pituinen altistus. Viisi lumekontrolloitua tutkimusta yhdistettiin (703 potilasta annoksella 100 mg kerran vuorokaudessa, 684 potilasta annoksella 200 mg kerran vuorokaudessa ja 438 potilasta lumelääkkeellä) abrositinibin turvallisuuden arvioimiseksi lumelääkkeeseen verrattuna enintään 16 viikon ajan.
Taulukossa 2 luetellaan haittavaikutukset, joita havaittiin atooppista ihottumaa koskevissa kliinisissä lääketutkimuksissa, esitettyinä elinjärjestelmän ja esiintymistiheyden mukaan, käyttäen seuraavia kategorioita: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin esiintymistiheysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 2. Haittavaikutukset | |||
| Elinjärjestelmäluokka | Hyvin yleinen | Yleinen | Melko harvinainen |
| Infektiot | Herpes simplexa Vyöruusu (Herpes zoster)b | Keuhkokuume | |
| Veri ja imukudos | Trombosytopenia Lymfopenia Neutropeniac | ||
| Aineenvaihdunta ja ravitsemus | Hyperlipidemiad | ||
| Hermosto | Päänsärky Heitehuimaus | ||
| Verisuonisto | Laskimotromboemboliae | ||
| Ruoansulatuselimistö | Pahoinvointi | Oksentelu Ylävatsakipu | |
| Iho ja ihonalainen kudos | Akne | ||
| Tutkimukset | Suurentunut kreatiinikinaasipitoisuus ˃ 5 × ULNf | ||
a. Herpes simplex sisältää seuraavat: suun herpesinfektio, silmänseudun Herpes simplex -infektio, genitaaliherpes ja herpesihottuma.
b. Herpes zoster sisältää silmänseudun vyöruusun.
c. Neutropenia sisältää pienentyneen neutrofiilien määrän ja granulosytopenian.
d. Hyperlipidemia sisältää dyslipidemian ja hyperkolesterolemian.
e. Laskimotromboembolia sisältää keuhkoembolian ja syvän laskimotukoksen.
f. Sisältää muutokset, jotka havaitaan laboratoriokokein tehtävässä seurannassa (ks. teksti alla).
Valikoitujen haittavaikutusten kuvaus
Infektiot
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa infektioita on raportoitu 27,4 %:lla lumelääkkeellä hoidetuista potilaista ja 34,9 %:lla abrositinibiannoksella 100 mg hoidetuista ja 34,8 %:lla Cibinqo-valmisteen annoksella 200 mg hoidetuista potilaista. Useimmat infektiot olivat lieviä tai keskivaikeita. Niiden potilaiden prosenttiosuus, jotka raportoivat infektioon liittyviä haittavaikutuksia 200 mg ja 100 mg -ryhmissä, lumelääkkeeseen verrattuna, olivat: Herpes simplex (4,2 % ja 2,8 % vs. 1,4 %), vyöruusu (Herpes zoster) (1,2 % ja 0,6 % vs. 0 %), keuhkokuume (0,1 % ja 0,1 % vs. 0 %). Herpes simplex oli yleisempi potilailla, joilla oli anamneesissa Herpes simplex tai Eczema herpeticum. Suurin osa Herpes zoster -tapahtumista sisälsi yksittäisen dermatomin eivätkä olleet vakavia. Suurin osa opportunistisista infektioista oli Herpes zoster ‑tapauksia (0,70 sataa potilasvuotta kohden 100 mg:n abrositinibiryhmässä ja 0,96 sataa potilasvuotta kohden 200 mg:n abrositinibiryhmässä), useimmat näistä olivat useita dermatomeja sisältäviä ihoinfektioita, jotka eivät olleet vakavia. Kliinisissä tutkimuksissa hoidetuilla potilailla, jotka saivat yhdenmukaisina hoito-ohjelmina joko 100 mg tai 200 mg abrositinibia, pitkäaikainen jatkotutkimus mukaan lukien, Herpes zoster -infektion ilmaantumistiheys abrositinibiannoksella 200 mg hoidetuilla potilailla (4,36 sataa potilasvuotta kohden) oli suurempi kuin annoksella 100 mg hoidetuilla potilailla (2,61 sataa potilasvuotta kohden). Herpes zoster ‑infektion ilmaantumistiheys oli suurempi myös vähintään 65‑vuotiailla potilailla (riskitiheyksien suhde 1,76) sekä potilailla, joilla oli anamneesissa vyöruusu (Herpes zoster) (riskitiheyksien suhde 3,41), lähtötilanteessa vaikea atooppinen ihottuma (riskitiheyksien suhde 1,17) ja ennen Herpes zoster ‑tapahtumaa varmistettu B-Lymf < 1,0 × 109/l (riskitiheyksien suhde 2,18) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa vakavien infektioiden määrä oli 1,81 sataa potilasvuotta kohden lumelääkkeellä hoidetuilla potilailla, 3,32 sataa potilasvuotta kohden potilailla, joita hoidettiin 100 mg:n annoksella, ja 1,12 sataa potilasvuotta kohden potilailla, joita hoidettiin 200 mg:n annoksella. Kaikkien kliinisissä tutkimuksissa yhdenmukaisina hoito-ohjelmina abrositinibiannoksella joko 100 mg tai 200 mg hoidettujen potilaiden joukossa, pitkäaikainen jatkotutkimus mukaan lukien, vakavien infektioiden määrä oli 2,20 sataa potilasvuotta kohden 100 mg:n annoksella hoidetuilla potilailla ja 2,48 sataa potilasvuotta kohden 200 mg:n annoksella hoidetuilla potilailla. Useimmin raportoidut vakavat infektiot olivat Herpes simplex, Herpes zoster ja keuhkokuume (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laskimotromboembolia
Kaikkien kliinisissä tutkimuksissa yhdenmukaisina hoito-ohjelmina abrositinibiannoksella joko 100 mg tai 200 mg hoidettujen potilaiden joukossa, pitkäaikainen jatkotutkimus mukaan lukien, keuhkoembolian esiintyvyys oli 0,21 sataa potilasvuotta kohden 200 mg:n annoksella ja 0,05 sataa potilasvuotta kohden 100 mg:n annoksella. Syvien laskimotukosten määrä oli 0,06 sataa potilasvuotta kohden 200 mg:n ryhmässä ja 0,05 sataa potilasvuotta kohden 100 mg:n ryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Trombosytopenia
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa hoitoon liittyi annosriippuvainen trombosyyttimäärien aleneminen. Maksimivaikutukset trombosyytteihin havaittiin 4 viikon sisällä, minkä jälkeen trombosyyttimäärä palautui kohti lähtötilannetta jatketusta hoidosta huolimatta. Vahvistetut trombosyyttimäärät < 50 × 109/l raportoitiin 0,1 %:lla potilaista, jotka altistuivat 200 mg:lle, ja 0 potilaalla, joita hoidettiin 100 mg:lla tai lumelääkkeellä. Kaikkien kliinisissä tutkimuksissa yhdenmukaisina hoito-ohjelmina abrositinibiannoksella joko 100 mg tai 200 mg hoidettujen potilaiden joukossa, pitkäaikainen jatkotutkimus mukaan lukien, vahvistettuja trombosyyttimääriä < 50 × 109/l raportoitiin 0,15 sataa potilasvuotta kohden 200 mg:lla hoidetuilla potilailla ja 0 sataa potilasvuotta kohden 100 mg:lla hoidetuilla potilailla, ja niitä todettiin useimmiten viikolla 4. Vähintään 65-vuotiailla potilailla todettiin yleisemmin trombosyyttimäärä < 75 × 109/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lymfopenia
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa vahvistettu absoluuttinen lymfosyyttimäärä < 0,5 × 109/l ilmeni 0,3 %:lla 200 mg:lla hoidetuista potilaista ja 0 %:lla 100 mg:lla tai lumelääkkeellä hoidetuista potilaista. Molemmat tapaukset ilmenivät ensimmäisten 4 altistusviikon aikana. Kaikkien kliinisissä tutkimuksissa yhdenmukaisina hoito-ohjelmina abrositinibiannoksella joko 100 mg tai 200 mg hoidettujen potilaiden joukossa, pitkäaikainen jatkotutkimus mukaan lukien, vahvistettuja absoluuttisia lymfosyyttimääriä < 0,5 × 109/l raportoitiin 0,34 sataa potilasvuotta kohden 200 mg:n annoksella ja 0,05 sataa potilasvuotta kohden 100 mg:n annoksella, ja niitä todettiin yleisemmin vähintään 65-vuotiailla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia
Kaikkien kliinisissä tutkimuksissa yhdenmukaisina hoito-ohjelmina abrositinibiannoksella joko 100 mg tai 200 mg hoidettujen potilaiden joukossa, pitkäaikainen jatkotutkimus mukaan lukien, vahvistettujen absoluuttisten neutrofiilimäärien < 1 × 109/l ilmaantuvuudeksi raportoitiin 0,03 sataa potilasvuotta kohden 200 mg:n annoksella ja 0 sataa potilasvuotta kohden 100 mg:n annoksella.
Lipidiarvojen nousut
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ilmeni annokseen liittyvä nousu LDL-kolesterolissa (LDL-c), kokonaiskolesterolissa, ja HDL-kolesterolissa (HDL-c) suhteessa lumelääkkeeseen viikolla 4. Arvot säilyivät koholla hoitojakson viimeiseen käyntiin asti. LDL/HDL-suhde ei muuttunut merkityksellisellä tavalla abrositinibilla hoidetuilla potilailla verrattuna lumelääkkeellä hoidettuihin potilaisiin. Hyperlipidemiaan liittyvät tapahtumat ilmenivät 0,4 %:lla 100 mg:n abrositinibiannokselle altistuneista potilaista, 0,6 %:lla 200 mg:n annokselle altistuneista potilaista ja 0 %:lla lumelääkkeelle altistuneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kreatiinikinaasiarvojen nousut (CK)
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa ilmeni merkittäviä kreatiinikinaasiarvojen nousuja (> 5 x ULN) 1,8 %:lla lumelääkkeellä hoidetuista potilaista, 1,8 %:lla 100 mg:lla hoidetuista potilaista ja 3,8 %:lla 200 mg:lla abrositinibia hoidetuista potilaista. Useimmat nousut olivat ohimeneviä eikä mikään niistä johtanut hoidon keskeyttämiseen.
Pahoinvointi
Enintään 16 viikkoa kestäneissä lumekontrolloiduissa tutkimuksissa pahoinvointia on raportoitu 1,8 %:lla lumelääkkeellä hoidetuista potilaista ja 6,3 %:lla Cibinqo-valmisteen annoksella 100 mg hoidetuista ja 15,1 %:lla Cibinqo-valmisteen annoksella 200 mg hoidetuista potilaista. Pahoinvointi johti hoidon keskeyttämiseen 0,4 %:lla abrositinibilla hoidetuista potilaista. Pahoinvoinnista kärsivistä potilaista 63,5 %:lla pahoinvointi alkoi ensimmäisen hoitoviikon aikana. Pahoinvoinnin mediaanikesto oli 15 vuorokautta. Useimmat tapauksista olivat vaikeusasteeltaan lieviä tai keskivaikeita.
Pediatriset potilaat
Kaikkiaan 635 nuorta iältään 12 – < 18-vuotiasta potilasta hoidettiin abrositinibilla atooppista ihottumaa koskevissa kliinisissä tutkimuksissa, mikä edustaa 1 326,1 altistuksen potilasvuotta. Nuorilla atooppista ihottumaa koskevissa tutkimuksissa havaittu turvallisuusprofiili oli samankaltainen kuin aikuispopulaatiossa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa Cibinqo-valmisteen suurin yksittäinen suun kautta annettu annos oli 800 mg ja 400 mg päivittäin 28 vuorokauden ajan. Haittavaikutukset olivat vertailukelpoisia pienemmillä annoksilla todettujen kanssa. Mitään spesifisiä toksisia vaikutuksia ei tunnistettu. Yliannoksen tapauksessa suositellaan, että potilasta tarkkaillaan haittavaikutusten merkkien ja oireiden varalta (ks. kohta Haittavaikutukset). Hoidon tulee olla oireenmukaista ja elintoimintoja tukevaa. Tämän lääkevalmisteen yliannokselle ei ole olemassa mitään spesifistä antidoottia.
Farmakokineettiset tiedot aina yksittäiseen suun kautta annettuun 800 mg:n annokseen asti aikuisilla terveillä vapaaehtoisilla osoittavat, että yli 90 %:n annetusta annoksesta odotetaan eliminoituvan 48 tunnin sisällä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut ihotautien lääkkeet, ihottumalääkkeet, lukuun ottamatta kortikosteroideja; ATC-koodi: D11AH08
Vaikutusmekanismi
Abrositinibi on Janus-kinaasin (JAK)1 estäjä. JAK:t ovat solunsisäisiä entsyymejä, jotka välittävät solukalvolla sytokiinin tai kasvutekijän ja sen reseptorin vuorovaikutuksesta syntyviä signaaleja ja vaikuttavat hematopoieesin ja immuunisolujen toiminnan solutason prosesseihin. JAK:t fosforyloivat ja aktivoivat Signal Transducers and Activators of Transcription (STAT) -tekijöitä, jotka muuntavat solunsisäistä aktiivisuutta, mm. geeniekspressiota. JAK1:n estäminen muuntaa signalointireittejä estämällä STAT-tekijöiden fosforylaatiota ja aktivaatiota.
Biokemiallisissa määrityksissä abrositinibilla on selektiivisyyttä JAK1:lle muiden kolmen JAK-isoformin suhteen, JAK2 (28‑kertainen), JAK3 (>340-kertainen) ja tyrosiinikinaasi 2 (TYK2, 43-kertainen). Solutasolla abrositinibi estää sytokiinien indusoimaa STAT:n fosforylaatiota JAK1:n sisältävien signalointiparien välityksellä, eikä sillä ole vaikutusta signalointiin JAK2/JAK2- tai JAK2/TYK2-parien välityksellä. Tiettyjen JAK-entsyymien selektiivisen entsyymitason estymisen merkitystä kliiniselle vaikutukselle ei tällä hetkellä tunneta.
Farmakodynaamiset vaikutukset
Kliiniset biomerkkiaineet
Abrositinibilla annettuun hoitoon liittyi annosriippuvainen seerumin tulehdusmerkkiaineiden määrän aleneminen atooppisessa ihottumassa [interleukiini-31 (IL‑31), interleukiini-22 (IL-22), eosinofiilimäärä, ja thymus and activation-regulated chemokine (TARC)], JAK1-signalointi [natural killer (NK) -solumäärä ja interferon gamma-induced protein 10 (IP-10)] tai molemmat [hyvin herkkä C-reaktiivinen proteiini (hsCRP)]. Nämä muutokset palautuivat hoidon lopettamisen jälkeen.
Keskimääräinen absoluuttinen lymfosyyttimäärä nousi 2 viikon jälkeen abrositinibihoidon aloittamisesta ja palasi lähtötasolle yhdeksänteen hoitokuukauteen mennessä. Useimmilla potilailla absoluuttinen lymfosyyttimäärä säilyi viitealueella. Abrositinibihoitoon liittyi annokseen liittyvä B-solujen määrän nousu ja annokseen liittyvä NK-solujen määrän lasku. Näiden B-solujen ja NK-solujen määrän muutosten kliinistä merkitystä ei tiedetä.
Sydämen elektrofysiologia
Abrositinibin vaikutusta QTc-aikaan selvitettiin tutkittavilla, jotka saivat yksittäisen supraterapeuttisen 600 mg:n annoksen abrositinibia lumelääkkeellä ja vaikuttavalla aineella kontrolloidussa QT-tutkimuksessa. Tutkimuksessa havaittiin abrositinibin pitoisuudesta riippuvainen QTc-aikaa pidentävä vaikutus; keskiarvo (90 %:n luottamusväli) QTc-ajan pitenemiselle oli 6,0 (4,52, 7,49) ms, mikä viittasi siihen, että abrositinibilla ei ole kliinisesti merkittävää vaikutusta QTc-aikaan testatulla annoksella.
Kliininen teho ja turvallisuus
Abrositinibin tehoa ja turvallisuutta monoterapiana ja yhdistelmänä taustalla olevien paikallislääkitysten kanssa arvioitiin 12–16 viikon ajan 1 616 potilaalla kolmessa keskeisessä vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (MONO-1, MONO‑2, ja COMPARE). Lisäksi abrositinibin tehoa ja turvallisuutta arvioitiin monoterapiana 52 viikon ajan (vaihtoehtona oli lisälääkkeen käyttö potilailla, joiden tauti paheni) 1 233 potilaalla, vaiheen 3 induktiohoitoa ja hoidon lopettamista koskevassa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (REGIMEN). Potilaat näissä neljässä tutkimuksessa olivat vähintään 12-vuotiaita, ja heillä oli keskivaikea tai vaikea atooppinen ihottuma, joka määriteltiin seuraavasti: IGA-mittarin (tutkijalääkärin tekemä yleinen arviointi, Investigator’s Global Assessment) pistemäärä ≥ 3, ihottuman pinta-alaa ja vaikeusastetta kuvaavan EASI-mittarin (Eczema Area and Severity Index) pistemäärä oli ≥ 16, ja ihottuma-alueen osuus kehon pinta-alasta (BSA) oli ≥ 10 %, ja pahinta kutinaa osoittavan numeerisen arviointiasteikon (Peak Pruritus Numerical Rating Scale, PP-NRS) pistemäärä oli ≥ 4 lähtötilanteessa ennen satunnaistamista. Potilaat, joilla oli aikaisempi riittämätön vaste tai joille paikalliset hoidot eivät olleet lääketieteellisesti suositeltavia, tai jotka olivat saaneet systeemisiä hoitoja, soveltuivat mukaan tutkimukseen. Kaikki potilaat, jotka olivat mukana kantatutkimuksessa sen loppuun saakka, soveltuivat jatkamaan pitkäaikaisessa EXTEND-jatkotutkimuksessa.
Lähtötilanteen ominaisuudet
Lumekontrolloiduissa tutkimuksissa (MONO-1, MONO-2, COMPARE) ja avoimessa tutkimuksessa (REGIMEN), jossa selvitettiin induktiohoitoa ja satunnaistettua hoidon lopettamista, kaikissa hoitoryhmissä 41,4 % – 51,1 % oli naisia, 59,3 % – 77,8 % oli valkoihoisia, 15,0 % – 33,0 % oli aasialaisia ja 4,1 % – 8,3 % oli mustaihoisia, ja keskimääräinen ikä oli 32,1–37,7 vuotta. Näihin tutkimuksiin otettiin mukaan kaikkiaan 134 potilasta, jotka olivat vähintään 65-vuotiaita. Näissä tutkimuksissa 32,2 % – 40,8 %:lla oli lähtötilanteessa IGA 4 (vaikea atooppinen ihottuma), ja 41,4 % – 59,5 % potilaista oli saanut aikaisemmin systeemistä hoitoa atooppiseen ihottumaan. Lähtötilanteen keskimääräinen EASI-pistemäärä oli 28,5–30,9, lähtötilanteen PP-NRS oli 7,0–7,3, ja lähtötilanteen DLQI (Dermatology Life Quality Index) oli 14,4–16,0.
Kliininen vaste
12 viikon monoterapiaa (MONO-1, MONO-2) ja 16 viikon yhdistelmähoitoa (COMPARE) koskevat -tutkimukset
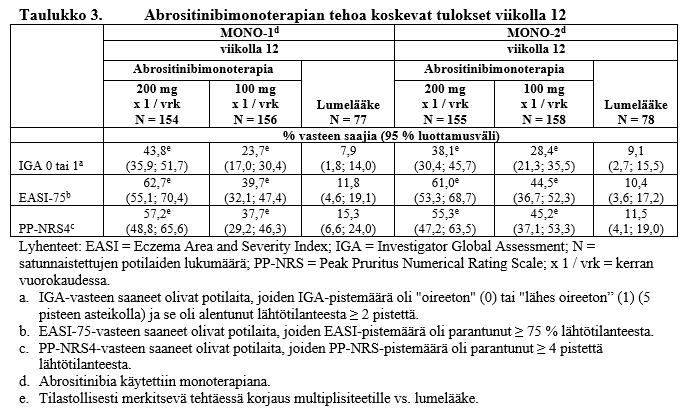
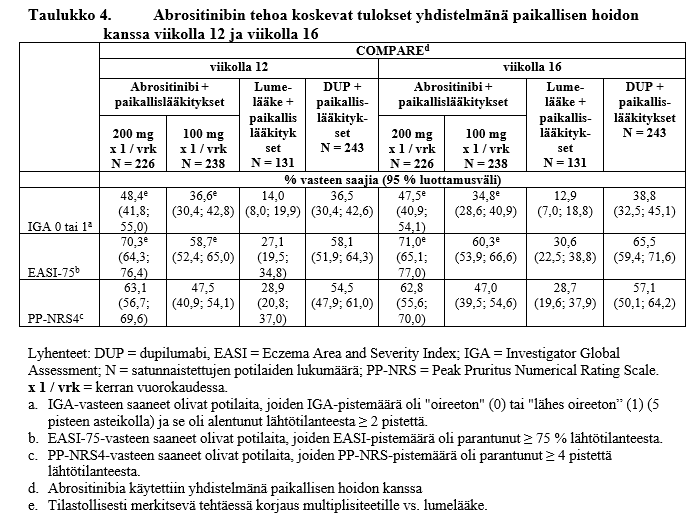
Merkitsevästi suurempi osuus potilaista saavutti molemmat ensisijaiset päätetapahtumat IGA 0 tai 1 ja/tai EASI‑75 kerran vuorokaudessa annetulla abrositinibiannoksella 100 mg tai 200 mg, lumelääkkeeseen verrattuna, viikolla 12 tai viikolla 16 (ks. taulukko 3 ja taulukko 4).
Merkitsevästi suurempi osuus potilaista saavutti ainakin PP-NRS:n 4 pisteen paranemisen kerran vuorokaudessa annetulla abrositinibiannoksella 100 mg tai 200 mg lumelääkkeeseen verrattuna. Tämä paraneminen havaittiin niinkin varhain kuin viikolla 2 ja se säilyi viikolle 12 (kuva 1).
COMPARE-tutkimuksessa abrositinibin 200 mg:n annoksen paremmuus verrattuna dupilumabiin viikolla 2 osoitettiin niiden potilaiden osuudessa, jotka saavuttivat ainakin PP-NRS:n 4 pisteen paranemisen merkitsevästi suuremmilla kutinavasteilla, jotka havaittiin niinkin varhain kuin päivänä 4 ensimmäisen annoksen jälkeen.
Hoitovaikutukset alaryhmissä (esim. paino, ikä, sukupuoli, rotu ja aikaisempi systeeminen immuunisalpaajahoito) tutkimuksissa MONO-1, MONO-2 ja COMPARE olivat yhdenmukaisia yleisessä tutkimuspopulaatiossa havaittujen tulosten kanssa.
Niiden potilaiden osuus, jotka saavuttivat ajan mittaan PP-NRS4:n tutkimuksissa MONO-1, MONO-2 ja COMPARE, esitetään kuvassa 1.
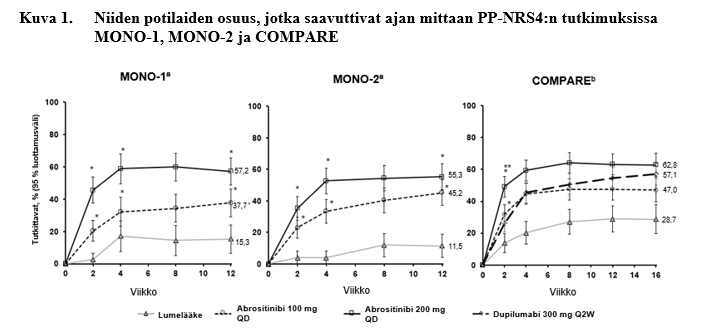
Lyhenteet: PP-NRS = Peak Pruritus Numerical Rating Scale; QD = kerran vuorokaudessa; Q2W = joka toinen viikko.
PP-NRS4-vasteen saaneet olivat potilaita, joiden PP-NRS-pistemäärä oli parantunut ≥ 4 pistettä lähtötilanteesta.
a. Abrositinibia käytettiin monoterapiana.
b. Abrositinibia käytettiin yhdistelmänä paikallisesti käytettävän lääkevalmisteen kanssa.
* Tilastollisesti merkitsevä tehtäessä korjaus multiplisiteetille vs. lumelääke.
** Tilastollisesti merkitsevä tehtäessä korjaus multiplisiteetille vs. dupilumabi.
Terveyteen liittyvät tulokset
Molemmissa monoterapiatutkimuksissa (MONO-1 ja MONO-2) ja yhdistelmähoitotutkimuksessa (COMPARE), abrositinibi paransi 12 viikon kohdalla merkitsevästi potilaiden ilmoittamia tuloksia, mukaan lukien kutina, uni (SCORAD Sleep VAS), AD-oireet (POEM), elämänlaatu (DLQI) ja ahdistuneisuus- ja masennusoireet (HADS) lumelääkkeeseen verrattuna. Tuloksia ei korjattu multiplisiteetin suhteen (ks. taulukko 5).
Taulukko 5. Potilaiden ilmoittamat tulokset abrositinibimonoterapiasta ja yhdistelmästä paikallisen hoidon kanssa viikolla 12
| Monoterapia | Yhdistelmähoito | ||||||||
| MONO-1 | MONO-2 | COMPARE | |||||||
200 mg x 1 / vrk | 100 mg x 1 / vrk | Lumelääke | 200 mg x 1 / vrk | 100 mg x 1 / vrk | Lumelääke | 200 mg x 1 / vrk + paikallis-lääkitykset | 100 mg x 1 / vrk + paikallis-lääkitykset | Lumelääke + paikallis-lääkitykset | |
| N | 154 | 156 | 77 | 155 | 158 | 78 | 226 | 238 | 131 |
SCORAD Sleep VAS, muutos lähtötilanteesta (95 %:n luottamusväli) | -3,7* (-4,2; -3,3) | -2,9* (-3,4; -2,5) | -1,6 (-2,2; -1,0) | -3,8* (-4,2; -3,4) | -3,0* (-3,4; -2,6) | -2,1 (-2,7; -1,5) | -4,6* (-4,9; -4,3) | -3,7* (-4,0; -3,4) | -2,4 (-2,8; -2,0) |
| DLQI ≥4 pisteen parantuminen, % vasteen saaneet | 72,6 %* | 67,2 %* | 43,6 % | 78,1 %* | 73,3 %* | 32,3 % | 86,4 %* | 74,7 %* | 56,5 % |
POEM, muutos lähtötilanteesta (95 %:n luottamusväli) | -10,6* (-11,8; -9,4) | -6,8* (-8,0; -5,6) | -3,7 (-5,5; -1,9) | -11,0* (-12,1; -9,8) | -8,7* (-9,9; -7,5) | -3,6 (-5,3; -1,9) | -12,6* (-13,6; -11,7) | -9,6* (-10,5; -8,6) | -5,1 (-6,3; -3,9) |
HADS Anxiety, muutos lähtötilanteesta (95 %:n luottamusväli) | -2,1* (-2,5; -1,6) | -1,6 (-2,0; -1,1) | -1,0 (-1,7; -0,4) | -1,7* (-2,2; -1,2) | -1,6* (-2,1; -1,1) | -0,6 (-1,3; 0,2) | -1,6* (-2,0; -1,2) | -1,2* (-1,5; -0,8) | -0,4 (-0,9; 0,1) |
HADS Depression, muutos lähtötilanteesta (95 %:n luottamusväli) | -1,8* (-2,2; -1,4) | -1,4* (-1,8; -0,9) | -0,2 (-0,8; 0,4) | -1,4* (-1,8; -1,0) | -1,0* (-1,5; -0,6) | 0,3 (-0,3; 0,9) | -1,6* (-1,9; -1,2) | -1,3* (-1,6; -0,9) | -0,3 (-0,7; 0,2) |
DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; N=satunnaistettujen potilaiden lukumäärä; POEM = Patient-Oriented Eczema Measure; x 1 / vrk =kerran vuorokaudessa; SCORAD = SCORing for AD; VAS=visual analog scale -jana
* Tilastollisesti merkitsevä ilman korjausta multiplisiteetille
Avoin induktiohoito- ja satunnaistettu hoidon lopettamistutkimus (REGIMEN)
Kaikkiaan 1 233 potilasta sai abrositinibia 200 mg kerran vuorokaudessa 12 viikon avoimessa esihoitovaiheessa. Näistä potilaista 798 (64,7 %) täytti vasteen saaneen kriteerit (määritelty seuraavasti: saavutti IGA [0 tai 1] -vasteen ja EASI‑75:n) ja satunnaistettiin saamaan lumelääkettä (267 potilasta), abrositinibia 100 mg kerran vuorokaudessa (265 potilasta) tai abrositinibia 200 mg kerran vuorokaudessa (266 potilasta).
Jatkuva hoito (200 mg jatkuva) esti taudin pahenemisjakson 81,1 %:n ja induktio-ylläpitohoito (200 mg 12 viikon ajan, jonka jälkeen 100 mg) 57,4 %:n todennäköisyydellä verrattuna 19,1 %:iin niiden potilaiden joukossa, jotka lopettivat hoidon (satunnaistettiin lumelääkeryhmään) 12 viikon induktion jälkeen. Kolmesataaviisikymmentäyksi (351) potilasta, joista 16,2 % oli saanut 200 mg:aa, 39,2 % oli saanut 100 mg:aa, ja 76,4 % oli saanut lumelääkettä, saivat varalääkkeenä 200 mg abrositinibia yhdistelmänä paikallislääkityksen kanssa.
| Kuva 2. Aika tutkimussuunnitelman määrittelemään pahenemisvaiheeseen |
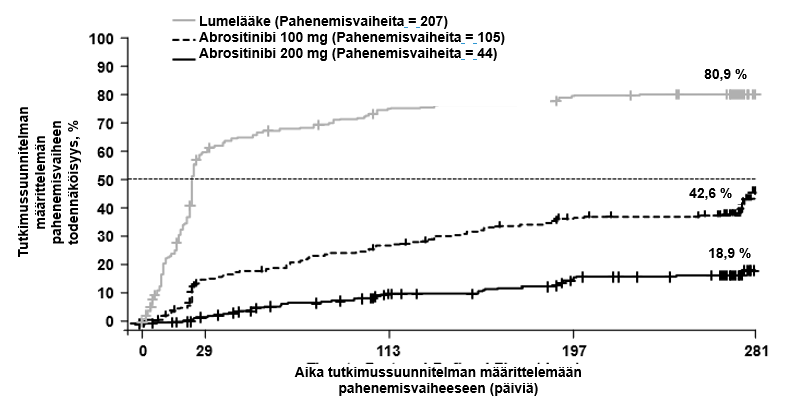 |
Abrositinibia käytettiin monoterapiana. Tutkimussuunnitelman määrittelemä pahenemisvaihe = Menetetään ainakin 50 % EASI-vasteesta viikolla 12 ja IGA‑pistemäärä ainakin 2. Multiplisiteettikontrolloitu p < 0,0001 200 mg vs. lumelääke; 100 mg vs. lumelääke; 200 mg vs. 100 mg. |
Pitkän aikavälin teho
Soveltuvia potilaita, jotka suorittivat loppuun täyden hoitojakson vaatimukset täyttävässä kantatutkimuksessa (esim. MONO-1, MONO‑2, COMPARE, REGIMEN), harkittiin rekrytoitaviksi pitkän aikavälin jatkotutkimukseen nimeltä EXTEND. EXTEND-tutkimuksessa potilaat saivat abrositinibia taustalla olevan paikallisesti käytettävän lääkehoidon kanssa tai ilman sitä. Potilaat, jotka satunnaistettiin aikaisemmin saamaan lääkevalmistetta 100 mg tai 200 mg kerran vuorokaudessa kantatutkimuksissa, jatkoivat samalla annoksella EXTEND-tutkimuksessa kuin kantatutkimuksessa. EXTEND-tutkimuksessa potilaat saivat kaksoissokkoutettua hoitoa, kunnes kantatutkimus suoritettiin loppuun, minkä jälkeen potilaat saivat yksöissokkoutettua hoitoa (hoitomääräys oli paljastettu tutkijoille mutta ei potilaille).
Niiden potilaiden joukossa, jotka saivat vasteen 12 hoitoviikon jälkeen ja siirtyivät EXTEND-tutkimukseen, suurin osa potilaista oli säilyttänyt vasteensa kumulatiivisen hoidon viikolla 96 molemmille abrositinibiannoksille [IGA (0 tai 1) -vaste: 64 % 100 mg kerran vuorokaudessa ja 72 % 200 mg kerran vuorokaudessa, EASI-75: 87 % 100 mg kerran vuorokaudessa ja 90 % 200 mg kerran vuorokaudessa, ja PP-NRS4: 100 mg kerran vuorokaudessa 75 % ja 200 mg kerran vuorokaudessa 80 %].
Niiden potilaiden joukossa, jotka eivät saaneet vastetta 12 hoitoviikon jälkeen ja siirtyivät EXTEND-tutkimukseen, osa potilaista sai myöhäisen vasteen jatkuvan abrositinibihoidon viikolla 24 (lähtötilanteesta laskien) [IGA (0 tai 1) -vaste: 25 % 100 mg kerran vuorokaudessa ja 29 % 200 mg kerran vuorokaudessa ja EASI-75: 50 % 100 mg kerran vuorokaudessa ja 200 mg kerran vuorokaudessa 57 %]. Potilaat, jotka saivat osittaisen vasteen viikolla 12, hyötyivät todennäköisemmin hoidosta viikolla 24 kuin ne potilaat, jotka eivät saaneet vastetta viikolla 12.
Potilaat, jotka saivat dupilumabia COMPARE-tutkimuksessa ja sittemmin siirtyivät EXTEND-tutkimukseen, satunnaistettiin saamaan joko 100 mg tai 200 mg abrositinibia kerran vuorokaudessa siirryttyään EXTEND-tutkimukseen. Niiden potilaiden joukossa, jotka eivät saaneet vastetta dupilumabille, huomattava osa potilaista sai vasteen 12 viikkoa sen jälkeen kun he olivat siirtyneet saamaan abrositinibia [IGA (0 tai 1) -vaste: 34 % 100 mg kerran vuorokaudessa ja 47 % 200 mg kerran vuorokaudessa, ja EASI-75: 100 mg kerran vuorokaudessa 68 % ja 200 mg kerran vuorokaudessa 80 %].
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset abrositinibin käytöstä atooppisen ihottuman hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
12 hoitoviikkoa kestäneessä abrositinibimonoterapiassa tehoa ja turvallisuutta arvioitiin kahdessa vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (MONO-1, MONO‑2), joissa oli mukana 124 iältään 12 – < 18-vuotiasta potilasta. Yli 52 hoitoviikkoa kestäneessä abrositinibimonoterapiassa (mahdollisuutena oli lisälääkkeen käyttö potilailla, joiden tauti paheni) tehoa ja turvallisuutta arvioitiin myös avoimessa induktiohoitoa ja satunnaistettua hoidon lopettamista koskevassa tutkimuksessa (REGIMEN), jossa oli mukana 246 iältään 12 – < 18-vuotiasta potilasta. Näissä tutkimuksissa tulokset nuorten alaryhmässä olivat yhdenmukaisia koko tutkimuspopulaatiolla saatujen tulosten kanssa.
Abrositinibin tehoa ja turvallisuutta yhdessä paikallisen lääkehoidon kanssa arvioitiin 12 hoitoviikkoa kestäneessä vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa TEEN-tutkimuksessa. Tutkimuksessa oli mukana 287 iältään 12 – < 18-vuotiasta potilasta, joilla oli keskivaikea tai vaikea atooppinen ihottuma määriteltynä seuraavasti: IGA-pistemäärä ≥ 3, EASI-pistemäärä ≥ 16, ihottuma-alueen osuus kehon pinta-alasta (BSA) ≥ 10 %, ja PP‑NRS ≥ 4 lähtötilannekäynnillä ennen satunnaistamista. Potilaat, joilla oli aikaisemmin riittämätön vaste tai jotka olivat saaneet systeemisiä hoitoja, soveltuivat mukaan tutkimukseen.
Lähtötilanteen ominaisuudet
TEEN-tutkimuksessa kaikissa hoitoryhmissä 49,1 % oli naisia, 56,1 % oli valkoihoisia, 33,0 % oli aasialaisia ja 6,0 % oli mustaihoisia potilaita. Mediaani-ikä oli 15 vuotta ja niiden potilaiden osuus, joilla oli vaikea atooppinen ihottuma (IGA 4), oli 38,6 %.
12 viikkoa kestäneen abrositinibihoidon tulokset nuorilla potilailla MONO-1- ja MONO-2- tutkimusten yhdistetyistä tiedoista ja TEEN-tutkimuksesta esitetään taulukossa 6.
| Taulukko 6. Tehoa koskevat tulokset nuorilla potilailla hoitoviikolla 12 MONO-1- ja MONO-2- tutkimusten yhdistetyistä tiedoista ja TEEN-tutkimuksesta | ||||||
| MONO-1 ja MONO-2 -tutkimusten yhdistetyt tiedot | TEENd | |||||
Abrositinibi 200 mg x 1 / vrk | Abrositinibi 100 mg x 1 / vrk | Lumelääke | Abrositinibi 200 mg x 1 / vrk | Abrositinibi 100 mg x 1 / vrk | Lumelääke | |
| IGA 0 tai 1a | ||||||
| N | 48 | 50 | 23 | 93 | 89 | 94 |
| % vasteen saajia | 31,3 | 22,0 | 8,7 | 46,2e | 41,6e | 24,5 |
| (95 %:n luottamusväli) | (18,1, 44,4) | (10,5, 33,5) | (0,0, 20,2) | (36,1, 56,4) | (31,3, 51,8) | (15,8, 33,2) |
| EASI-75b | ||||||
| N | 48 | 50 | 23 | 93 | 89 | 94 |
| % vasteen saajia | 56,3 | 44,0 | 8,7 | 72,0e | 68,5e | 41,5 |
| (95 %:n luottamusväli) | (42,2, 70,3) | (30,2, 57,8) | (0,0, 20,2) | (62,9, 81,2) | (58,9, 78,2) | (31,5, 51,4) |
| PP-NRS4c | ||||||
| N | 36 | 42 | 22 | 74 | 76 | 84 |
| % vasteen saajia | 61,1 | 28,6 | 9,1 | 55,4e | 52,6 | 29,8 |
| (95 %:n luottamusväli) | (45,2, 77,0) | (14,9, 42,2) | (0,0, 21,1) | (44,1, 66,7) | (41,4, 63,9) | (20,0, 39,5) |
Lyhenteet: EASI = Eczema Area and Severity Index; IGA = Investigator Global Assessment; N = arvioitavissa olevien potilaiden lukumäärä; PP-NRS = Peak Pruritus Numerical Rating Scale; x 1 / vrk = kerran vuorokaudessa. a. IGA-vasteen saaneet olivat potilaita, joiden IGA-pistemäärä oli "oireeton" (0) tai "lähes oireeton” (1) (5 pisteen asteikolla) ja se oli alentunut lähtötilanteesta ≥ 2 pistettä. b. EASI-75-vasteen saaneet olivat potilaita, joiden EASI-pistemäärä oli parantunut ≥ 75 % lähtötilanteesta. c. PP-NRS4-vasteen saaneet olivat potilaita, joiden PP-NRS-pistemäärä oli parantunut ≥ 4 pistettä lähtötilanteesta. d. Abrositinibia käytettiin yhdistelmänä paikallisen lääkehoidon kanssa. e. Tilastollisesti merkitsevä tehtäessä korjaus multiplisiteetille vs. lumelääke. | ||||||
Niiden nuorten potilaiden joukossa, jotka saivat vasteen 12 hoitoviikon jälkeen ja siirtyivät pitkäaikaiseen EXTEND-jatkotutkimukseen, suurimmalla osalla potilaista vaste oli säilynyt kumulatiivisen hoidon viikolla 96 molemmilla abrositinibiannoksilla [100 mg ja 200 mg kerran vuorokaudessa: IGA (0 tai 1) ‑vaste 62 % ja 78 %, EASI‑75 89 % ja 93 %, PP-NRS4 77 % ja 76 %].
Niiden nuorten potilaiden joukossa, jotka eivät saaneet vastetta 12 hoitoviikon jälkeen ja siirtyivät EXTEND-tutkimukseen, osa potilaista sai myöhäisen vasteen jatkuvan abrositinibihoidon viikkoon 24 mennessä (lähtötilanteesta laskien) molemmilla abrositinibiannoksilla [100 mg ja 200 mg kerran vuorokaudessa: IGA (0 tai 1) ‑vaste 34 % ja 28 %, EASI‑75 41 % ja 55 %].
Farmakokinetiikka
Imeytyminen
Abrositinibi imeytyy hyvin, suun kautta annetusta lääkeaineesta imeytyy yli 91 %, ja abrositinibin oraalinen absoluuttinen biologinen hyötyosuus on noin 60 %. Suun kautta annetun abrositinibin imeytyminen on nopeaa ja plasman huippukonsentraatiot saavutetaan tunnissa. Abrositinibin vakaan tilan plasmakonsentraatio saavutetaan 48 tunnin kuluessa kerran vuorokaudessa annon jälkeen. Abrositinibin sekä Cmax että AUC lisääntyivät suhteessa annokseen enintään 200 mg:aan asti. Abrositinibin antamisella runsasrasvaisen aterian kanssa ei ollut kliinisesti merkittävää vaikutusta abrositinibialtistukseen (AUC suureni noin 26 % ja Cmax suureni noin 29 %, ja Tmax piteni kahdella tunnilla). Kliinisissä tutkimuksissa abrositinibi annettiin aterioista riippumatta (ks. kohta Annostus ja antotapa).
Jakautuminen
Laskimoon tapahtuneen annon jälkeen abrositinibin jakautumistilavuus on noin 100 litraa. Noin 64 % verenkierrossa olevasta abrositinibista, 37 % aktiivisesta metaboliitista M1 ja 29 % aktiivisesta metaboliitista M2 sitoutuvat plasman proteiineihin. Abrositinibi ja sen aktiiviset metaboliitit jakautuvat yhtäläisesti punasolujen ja plasman välille.
Biotransformaatio
Abrositinibin in vitro -metaboliaa välittävät useat CYP-entsyymit, CYP2C19 (~53 %), CYP2C9 (~30 %), CYP3A4 (~11 %) ja CYP2B6 (~6 %). Ihmisillä suoritetuissa radioleimatussa tutkimuksessa abrositinibi oli verenkierron vallitsevin muoto, lähinnä 3 poolista monohydroksyloitunutta metaboliittia tunnistettiin seuraaviksi: M1 (3‑hydroksipropyyli), M2 (2-hydroksipropyyli) ja M4 (pyrrolidinonipyrimidiini). Vakaassa tilassa M2 ja M4 ovat tärkeimmät metaboliitit ja M1 on vähäinen metaboliitti. Kolmesta verenkierrossa olevasta metaboliitista M1:lla ja M2:lla on samanlaiset JAK:ia estävät profiilit kuin abrositinibilla, kun taas M4 oli farmakologisesti inaktiivinen. Abrositinibin farmakologinen aktiivisuus liittyy altistumiseen sitoutumattomalle kantamolekyylille (~60 %) sekä M1:lle (~10 %) ja M2:lle (~30 %) systeemisessä verenkierrossa. Abrositinibin aktiiviseksi ainesosaksi kutsutaan seuraavien summaa: altistumiset sitoutumattomalle abrositinibille, M1:lle ja M2:lle, joista kukin ilmaistaan mooliyksikköinä ja sovitetaan suhteellisten vahvuuksien suhteen.
Yhteisvaikutustutkimuksissa BCRP:n ja OAT3:n (esim. rosuvastatiini), MATE1/2K:n (esim. metformiini), CYP3A4:n (esim. midatsolaami) ja CYP2B6:n (esim. efavirentsi) substraattien kanssa abrositinibilla ei havaittu olevan kliinisesti merkittävää vaikutusta.
Eliminaatio
Abrositinibin eliminaation puoliintumisaika on noin 5 tuntia. Abrositinibi eliminoituu pääasiallisesti metabolisten puhdistumamekanismien kautta, ja alle 1 % annoksesta erittyy virtsaan muuttumattomana vaikuttavana aineena. Abrositinibin metaboliitit, M1, M2 ja M4, erittyvät pääasiallisesti virtsaan ja ovat OAT3-kuljetusproteiinin substraatteja.
Erityiset potilasryhmät
Paino, sukupuoli, genotyyppi, rotu ja ikä
Painolla, sukupuolella, CYP2C19/2C9-genotyypillä, rodulla ja iällä ei ollut kliinisesti merkittävää vaikutusta abrositinibialtistukseen (ks. kohta Annostus ja antotapa).
Nuoret (≥ 12 – < 18 vuotta)
Populaatiofarmakokineettisen analyysin perusteella keskimääräisissä vakaan tilan abrositinibialtistuksissa ei ollut kliinisesti merkittävää eroa normaalipainoisten nuorten ja aikuisten välillä.
Lapset (< 12 vuotta)
Yhteisvaikutuksia on tutkittu vain aikuisilla. Abrositinibin farmakokinetiikkaa alle 12-vuotiailla lapsilla ei ole varmistettu (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa koskevassa tutkimuksessa potilailla, joilla oli vaikea (eGFR < 30 ml/min) ja keskivaikea (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta, aktiivisen rakenneosan AUCinf lisääntyi vaikeissa tapauksissa noin 191 % ja keskivaikeissa tapauksissa noin 110 %, verrattuna potilaisiin, joiden munuaiset toimivat normaalisti (eGFR ≥ 90 ml/min) (ks. kohta Annostus ja antotapa). Abrositinibin farmakokinetiikkaa ei ole määritetty potilailla, joilla on lievä munuaisten vajaatoiminta; muilla ryhmillä havaittujen tulosten perusteella on kuitenkin odotettavissa jopa 70 %:n lisäys aktiiviselle rakenneosalle altistumiselle potilailla, joilla on lievä munuaisten vajaatoiminta (eGFR 60 – < 90 ml/min). Jopa 70 %:n lisäys ei ole kliinisesti merkittävä, koska abrositinibin teho ja turvallisuus potilailla, joilla oli atooppinen ihottuma ja lievä munuaisten vajaatoiminta (n = 756) oli vertailukelpoinen yleisen populaation kanssa vaiheen 2 ja 3 kliinisissä tutkimuksissa. Yksittäisten potilaiden eGFR-arvot arvioitiin käyttämällä Modification of Diet in Renal Disease (MDRD) -yhtälöä.
Abrositinibia ei ole tutkittu ESRD-potilailla, jotka saavat munuaiskorvaushoitoa (ks. kohta Annostus ja antotapa). Vaiheen 3 kliinisissä lääketutkimuksissa abrositinibia ei arvioitu potilailla, joilla oli atooppinen ihottuma ja joiden kreatiniinipuhdistuma-arvot olivat lähtötilanteessa alle 40 ml/min.
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (Child Pugh A) sairastavien potilaiden aktiivisen rakenneosan AUCinf aleni noin 4 % ja keskivaikeaa maksan vajaatoimintaa (Child Pugh B) sairastavien potilaiden aktiivisen rakenneosan AUCinf suureni 15 %, verrattuna potilaisiin, joiden maksa toimi normaalisti. Nämä muutokset eivät ole kliinisesti merkittäviä eikä annosta tarvitse muuttaa potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Kliinisissä lääketutkimuksissa abrositinibia ei tutkittu potilailla, joilla oli vaikea (Child Pugh C) maksan vajaatoiminta (ks. kohta Vasta-aiheet), tai potilailla, joilla oli seulonnassa havaittu aktiivinen B-hepatiitti tai C‑hepatiitti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Prekliiniset tiedot turvallisuudesta
Yleinen toksisuus
Prekliinisissä tutkimuksissa havaittiin alentuneita lymfosyyttiarvoja ja immuuni- ja hematopoieettisten järjestelmien elinten/kudosten koon pienentymistä ja/tai lymfoidisia soluja. Näiden katsottiin liittyvän abrositinibin farmakologisiin ominaisuuksiin (JAK-kinaasin esto).
Toksisuustutkimuksissa rotille, joiden ikä vastasi vähintään 12-vuotiaan nuoren ihmisen ikää, annettiin enintään yhden kuukauden ajan abrositinibia. Tutkimuksissa havaittiin mikroskooppinen luudystrofialöydös, jota pidettiin ohimenevänä ja korjautuvana, ja altistumismarginaalit, joissa luulöydöstä ei havaittu, olivat 5,7–6,1 kertaa ihmisen AUC suurimmalla suositellulla ihmisen annoksella (MRHD) 200 mg. Mitään luulöydöksiä ei havaittu rotilla millään annoksella 6 kuukauden pituisessa toksisuustutkimuksessa (enintään 25 kertaa ihmisen AUC MRHD-annoksella 200 mg) tai missään jaavanmakakeilla tehdyissä toksisuustutkimuksissa (vastaa ihmisen ikää vähintään 8 vuotta; enintään 30 kertaa ihmisen AUC MRHD-annoksella 200 mg).
Genotoksisuus
Abrositinibi ei ollut mutageeninen bakteerimutageenisuusmäärityksessä (Amesin testi). Se ei ollut aneugeeninen tai klastogeeninen rotan luuytimen in vivo mikrotumamäärityksen perusteella.
Karsinogeenisuus
Mitään näyttöä tuumorigeenisuudesta ei havaittu 6 kuukauden ikäisillä Tg.rasH2-hiirillä, joille annettiin abrositinibia suun kautta enintään 75 mg/kg/vrk (naarashiiret) ja 60 mg/kg/vrk (uroshiiret). Kahden vuoden pituisessa karsinogeenisuustutkimuksessa naarasrotilla havaittiin enemmän hyvänlaatuisia tymoomia pienimmällä testatulla annoksella. Näin ollen LOAEL-arvo (pienin havaittavan haittavaikutuksen aiheuttava annos) asetetaan naaraille altistumisilla, jotka vastaavat arvoa 0,6 kertaa ihmisen AUC MRHD-arvolla 200 mg. Uroksille NOAEL-arvo (suurin annos, joka ei aiheuta haittavaikutuksia) asetettiin altistumisilla, jotka vastasivat arvoa 13 kertaa ihmisen AUC MRHD-arvolla 200 mg. Hyvänlaatuisen tymooman merkitystä ihmisille ei tunneta.
Lisääntymis- ja kehitystoksisuus
Abrositinibilla ei ollut vaikutusta koiraiden hedelmällisyyteen tai spermatogeneesiin. Abrositinibilla oli vaikutuksia naaraiden hedelmällisyyteen (pienempi hedelmällisyysindeksi, corpora lutea, implantaatiokohdat ja implantaation jälkeen tapahtuva alkion menetys), mutta mitään vaikutuksia hedelmällisyyteen ei havaittu altistuksilla, jotka vastasivat arvoa 1,9 kertaa ihmisen AUC MRHD-arvolla 200 mg. Vaikutukset korjautuivat yhden kuukauden kuluttua hoidon lopettamisesta.
Mitään sikiön epämuodostumia ei havaittu embryofetaalisissa kehitystutkimuksissa rotilla tai kaniineilla. Tiineillä kaniineilla suoritetussa embryofetaalisessa kehitystutkimuksessa vaikutuksia embryofetaaliseen eloonjäämiseen havaittiin pienimmällä testatulla annoksella, altistukset vastasivat arvoa 0,14 kertaa sitoutumaton ihmisen AUC MRHD-arvolla 200 mg. Poikueissa havaittiin enemmän tapauksia, joissa takaraajan varpaat ja nilkkaluut ja eturaajan varpaat eivät olleet luutuneet. Näitä havaittiin altistuksilla, jotka vastasivat arvoa 0,14 kertaa sitoutumaton ihmisen AUC MRHD-arvolla 200 mg.
Vaikka tiineillä rotilla suoritetussa embryofetaalisessa kehitystutkimuksessa havaittiinkin enemmän embryofetaalista letaalisuutta, mitään sellaista ei havaittu altistuksilla, jotka vastasivat arvoa 10 kertaa ihmisen AUC MRHD-arvolla 200 mg. Sikiöillä havaittiin enemmän luustomuutoksia: lyhyet 13. kylkiluut, pienemmät ventraaliset haarakkeet, paksuuntuneet kylkiluut, ja luutumattomat jalkapöytäluut, mutta mitään näistä ei havaittu altistuksilla, jotka vastasivat arvoa 2,3 kertaa ihmisen AUC MRHD-arvolla 200 mg.
Tiineillä rotilla suoritetussa pre- ja postnataalista kehitystä koskevassa tutkimuksessa emoilla oli synnytysvaikeuksia ja synnytys kesti pitempään, poikasten painot olivat pienempiä ja postnataalinen eloonjääminen oli harvinaisempaa. Mitään emoon kohdistuvaa toksisuutta tai kehitystoksisuutta ei havaittu emoilla tai poikasilla altistuksilla, jotka vastasivat arvoa 2,3 kertaa ihmisen AUC MRHD‑annoksella 200 mg.
Abrositinibin anto nuorille rotille postnataalisesta päivästä 10 alkaen (vastasivat 3 kuukauden ikäistä ihmistä) johti haitallisiin mikroskooppisiin ja makroskooppisiin luulöydöksiin, joita olivat väärin kiertyneet tassut, murtumat ja/tai reisiluun pään epänormaalit tilat. Näitä havaittiin altistuksilla, jotka vastasivat arvoa ≥ 0,8 kertaa ihmisen AUC MRHD-arvolla 200 mg. Kun abrositinibin anto nuorille rotille aloitettiin postnataalisena päivänä 21 tai sen jälkeen (vastasivat vähintään 2 vuoden ikäistä ihmistä), siihen ei liittynyt mikroskooppisia eikä makroskooppisia luulöydöksiä.
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Mikrokiteinen selluloosa (E460i)
Kalsiumvetyfosfaatti vedetön (E341ii)
Natriumtärkkelysglykolaatti
Magnesiumstearaatti (E470b)
Kalvopäällyste
Hypromelloosi (E464)
Titaanidioksidi (E171)
Laktoosimonohydraatti
Makrogoli (E1521)
Triasetiini (E1518)
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CIBINQO tabletti, kalvopäällysteinen
50 mg (L:kyllä) 28 fol (868,21 €)
100 mg (L:kyllä) 28 fol (868,21 €)
200 mg (L:kyllä) 28 fol (868,21 €)
PF-selosteen tieto
Cibinqo 50 mg kalvopäällysteiset tabletit
Suuritiheyksinen polyeteeni (HDPE) -purkki ja polypropeenikorkki, sisältää 14 tai 30 kalvopäällysteistä tablettia.
Polyvinylideenikloridi (PVDC) -läpipainopakkaus, jossa alumiinikalvokansi, sisältää 7 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 14, 28 tai 91 kalvopäällysteistä tablettia.
Cibinqo 100 mg kalvopäällysteiset tabletit
HDPE-purkki ja polypropeenikorkki, sisältää 14 tai 30 kalvopäällysteistä tablettia.
PVDC-läpipainopakkaus, jossa alumiinikalvokansi, sisältää 7 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 14, 28 tai 91 kalvopäällysteistä tablettia.
Cibinqo 200 mg kalvopäällysteiset tabletit
HDPE-purkki ja polypropeenikorkki, sisältää 14 tai 30 kalvopäällysteistä tablettia.
PVDC-läpipainopakkaus, jossa alumiinikalvokansi, sisältää 7 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 14, 28 tai 91 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Cibinqo 50 mg kalvopäällysteiset tabletit
Vaaleanpunainen, noin 11 mm pitkä ja 5 mm leveä soikionmuotoinen tabletti, jonka toiselle puolelle on kaiverrettu ”PFE” ja vastakkaiselle puolelle ”ABR 50”.
Cibinqo 100 mg kalvopäällysteiset tabletit
Vaaleanpunainen, halkaisijaltaan noin 9 mm pyöreä tabletti, jonka toiselle puolelle on kaiverrettu ”PFE” ja vastakkaiselle puolelle ”ABR 100”.
Cibinqo 200 mg kalvopäällysteiset tabletit
Vaaleanpunainen, noin 18 mm pitkä ja 8 mm leveä soikionmuotoinen tabletti, jonka toiselle puolelle on kaiverrettu ”PFE” ja vastakkaiselle puolelle ”ABR 200”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CIBINQO tabletti, kalvopäällysteinen
50 mg 28 fol
100 mg 28 fol
200 mg 28 fol
- Ylempi erityiskorvaus (100 %). Abrositinibi ja upadasitinibi (vaikea atooppinen ihottuma): Vaikeaan atooppiseen ihottumaan liittyvän yleisen erytrodermian hoito erityisin edellytyksin (1543).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abrositinibi: Aikuisten ja vähintään 12-vuotiaiden nuorten vaikean atooppisen ihottuman hoito erityisin edellytyksin (3073).
ATC-koodi
D11AH08
Valmisteyhteenvedon muuttamispäivämäärä
23.06.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com