

SUNITINIB KRKA kapseli, kova 25 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää sunitinibimalaattia määrä, joka vastaa 12,5 mg:aa sunitinibia.
Yksi kova kapseli sisältää sunitinibimalaattia määrä, joka vastaa 25 mg:aa sunitinibia.
Yksi kova kapseli sisältää sunitinibimalaattia määrä, joka vastaa 50 mg:aa sunitinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli (kapseli).
Kliiniset tiedot
Käyttöaiheet
Gastrointestinaalinen stroomakasvain (GIST)
Sunitinib Krka on tarkoitettu inoperaabelin ja/tai metastasoituneen pahanlaatuisen gastrointestinaalisen stroomakasvaimen (GIST) hoitoon aikuisille, silloin kun imatinibihoito on resistenssin tai intoleranssin vuoksi epäonnistunut.
Metastasoitunut munuaissolukarsinooma (MRCC)
Sunitinib Krka on tarkoitettu edenneen/metastasoituneen munuaissolukarsinooman (MRCC) hoitoon aikuisille.
Haiman neuroendokriiniset kasvaimet (haiman NET)
Sunitinib Krka on tarkoitettu inoperaabeleiden tai metastasoituneiden hyvin erilaistuneiden haiman neuroendokriinisten kasvainten (haiman NET) hoitoon aikuisille taudin edetessä.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Sunitinib Krka -hoidon aloittavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus
GIST ja MRCC: Sunitinib Krka -valmisteen suositusannostus on 50 mg kerran vuorokaudessa suun kautta 4 perättäisen viikon ajan, minkä jälkeen on 2 viikon hoitotauko (hoito-ohjelma 4/2). Tästä muodostuu yksi 6 viikon hoitosykli.
Haiman NET: Sunitinib Krka -valmisteen suositusannos on 37,5 mg kerran vuorokaudessa suun kautta ilman suunniteltua hoitotaukoa.
Annosmuutokset
Turvallisuus ja siedettävyys
GIST ja MRCC: Annosta voidaan muuttaa 12,5 mg:n kerta-annoksin potilaan yksilöllisen turvallisuuden ja sietokyvyn perusteella. Vuorokausiannos ei saa olla yli 75 mg eikä alle 25 mg.
Haiman NET: Annosta voidaan muuttaa 12,5 mg kerta-annoksin potilaan yksilöllisen turvallisuuden ja sietokyvyn perusteella. Kolmannen vaiheen haiman NET -tutkimuksessa suurin käytetty annos oli
50 mg vuorokaudessa.
Annostelun keskeyttäminen voi olla tarpeen potilaan yksilöllisen turvallisuuden ja sietokyvyn vuoksi.
CYP3A4:n estäjät/induktorit
Samanaikaista potenttien CYP3A4:n induktorien (kuten rifampisiinin) antoa on vältettävä (ks. kohdat
4.4 ja 4.5). Jos tämä ei ole mahdollista, sunitinibin annosta on ehkä suurennettava 12,5 mg:n lisäyksin (enintään kokonaisannokseen 87,5 mg/vrk GIST- ja MRCC-käyttöaiheissa tai enintään kokonaisannokseen 62,5 mg/vrk haiman NET -käyttöaiheeessa) siedettävyyden huolellisen seurannan perusteella.
Sunitinibin samanaikaista antoa potenttien CYP3A4:n estäjien (kuten ketokonatsolin) kanssa on vältettävä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset). Jos tämä ei ole mahdollista, sunitinibin annos on ehkä pienennettävä vähimmäismäärään 37,5 mg/vrk GIST- ja MRCC-käyttöaiheissa tai vähimmäismäärään 25 mg/vrk haiman NET -käyttöaiheessa siedettävyyden huolellisen seurannan perusteella.
Potilaalle on harkittava jotakin sellaista samanaikaista vaihtoehtoista lääkevalmistetta, jonka ei odoteta indusoivan tai estävän CYP3A4:ää lainkaan tai vain vähän.
Erityisryhmät
Pediatriset potilaat
Sunitinib Krka -valmisteen turvallisuutta ja tehoa alle 18-vuotiaille potilaille ei ole osoitettu.
Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Iäkkäät
Kliinisissä tutkimuksissa noin 1⁄3 sunitinibia saaneista potilaista oli vähintään 65-vuotiaita. Nuorten ja iäkkäiden potilaiden välillä ei havaittu merkittäviä eroja turvallisuudessa tai hoidon tehossa.
Maksan vajaatoiminta
Aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta (Child-Pughin luokat A ja B). Sunitinibia ei ole tutkittu tutkimushenkilöillä, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokka C), ja siksi sen käyttöä ei voida suositella potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Aloitusannoksen muutosta ei vaadita annosteltaessa sunitinibia potilaille, joilla on munuaisten vajaatoiminta (lievästä vaikeaan) tai dialyysihoitoa vaativa loppuvaiheen munuaissairaus. Annosmuutosten tulee perustua potilaan yksilölliseen turvallisuuteen ja sietokykyyn (ks. kohta Farmakokinetiikka).
Antotapa
Sunitinib Krka otetaan suun kautta joko ruoan kanssa tai ilman ruokaa.
Jos annos jää väliin, potilaan ei pidä ottaa ylimääräistä annosta. Potilaan tulee ottaa tavanomainen lääkärin määräämä annos seuraavana päivänä.
Sunitinibi-kapseleita ei saa painaa läpipainopakkauksen folion läpi, sillä kapseli voi rikkoutua. Kapseli tulee poistaa pakkauksesta vetämällä folio irti yhdestä irrotetusta kapselilokerosta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Samanaikaista antoa potenttien CYP3A4:n induktorien kanssa on vältettävä, koska se voi pienentää plasman sunitinibipitoisuutta (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Samanaikaista antoa potenttien CYP3A4:n estäjien kanssa on vältettävä, koska se voi suurentaa plasman sunitinibipitoisuutta (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Ihon ja kudosten häiriöt
Potilaalle on kerrottava, että sunitinibihoidon aikana voi ilmetä hiusten tai ihon depigmentaatiota. Muita mahdollisia ihovaikutuksia ovat ihon kuivuminen, paksuneminen tai halkeileminen, rakkulamuodostus tai ihottuma kämmenissä ja jalkapohjissa.
Nämä vaikutukset eivät olleet kumulatiivisia vaan tyypillisesti korjaantuivat eivätkä yleensä johtaneet hoidon lopettamiseen. Pyoderma gangrenosum -tapauksia on raportoitu. Nämä tapaukset yleensä paranivat, kun sunitinibin anto keskeytettiin. Vakavia ihoreaktioita, mm. erythema multiformea sekä Stevens-Johnsonin oireyhtymään ja toksiseen epidermaaliseen nekrolyysiin viittaavia tapauksia, on raportoitu. Osa näistä tapauksista on johtanut kuolemaan. Jos näihin sairauksiin viittaavia merkkejä tai oireita ilmenee (esim. paheneva ihottuma, johon liittyy rakkuloita tai limakalvovaurioita), sunitinibihoito tulee keskeyttää. Jos Stevens-Johnsonin oireyhtymän tai toksisen epidermaalisen nekrolyysin diagnoosi varmistuu, hoitoa ei saa aloittaa uudelleen. Joissakin epäillyissä erythema multiformen tapauksissa potilaat sietivät sunitinibihoidon aloittamisen uudelleen pienemmällä annoksella reaktion häviämisen jälkeen. Osa näistä potilaista sai myös samanaikaista hoitoa kortikosteroideilla tai antihistamiineilla (ks. kohta Haittavaikutukset).
Verenvuoto ja kasvaimen verenvuoto
Sunitinibilla tehdyissä kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeisessä seurannassa on ilmoitettu verenvuototapahtumia, kuten verenvuotoja maha-suolikanavassa, hengitysteissä, virtsateissä ja aivoissa. Osa näistä tapahtumista on johtanut kuolemaan (ks. kohta Haittavaikutukset).
Verenvuototapahtumien tavanomaiseen arviointiin tulee kuulua täydellinen verenkuva ja lääkärintarkastus.
Verenvuotohaittavaikutuksista yleisin oli nenäverenvuoto, jota ilmoitettiin noin puolella niistä potilaista, joilla oli kiinteitä kasvaimia ja joilla oli ilmennyt jokin verenvuototapahtuma. Jotkut nenäverenvuototapahtumista olivat vaikeita, mutta johtivat kuolemaan vain erittäin harvoin.
Kasvaimen verenvuotoa, joskus liittyen kasvaimen nekroosiin, on raportoitu. Jotkut näistä verenvuototapahtumista johtivat kuolemaan.
Verenvuoto kasvaimessa voi alkaa yhtäkkiä. Jos kyseessä on keuhkokasvain, verenvuoto kasvaimesta voi ilmetä vaikeana ja hengenvaarallisena veriyskänä (hemoptyysi) tai verenvuotona keuhkoista. Keuhkoverenvuotoa on esiintynyt kliinisissä tutkimuksissa, ja sitä on raportoitu myös markkinoille tulon jälkeen sunitinibihoitoa saaneilla MRCC-, GIST- ja keuhkosyöpäpotilailla. Osa tapauksista on johtanut kuolemaan. Sunitinibia ei ole hyväksytty keuhkosyövän hoitoon.
Jos potilas saa samanaikaisesti verenhyytymistä estävää lääkehoitoa (esim. varfariinia, asenokumarolia), häneltä on aika ajoin määritettävä täydellinen verenkuva (trombosyytit) ja hyytymistekijät (P-TT-INR), ja hänelle on tehtävä lääkärintarkastus.
Ruoansulatuskanavan häiriöt
Yleisimmin ilmoitetut ruoansulatuskanavan haittavaikutukset olivat ripuli, pahoinvointi/oksentelu, vatsakipu, dyspepsia ja stomatiitti/suukipu. Myös esofagiittia on raportoitu (ks. kohta Haittavaikutukset).
Ruoansulatuskanavan haittavaikutusten hoitoon voi kuulua pahoinvointi-, ripuli- tai antasidilääkitys.
Joissakin tapauksissa niillä sunitinibilla hoidetuilla potilailla, joilla oli vatsaontelon pahanlaatuinen kasvain, raportoitiin vakavia, jopa kuolemaan johtaneita ruoansulatuskanavan komplikaatioita, mukaan lukien ruoansulatuskanavan puhkeama.
Kohonnut verenpaine
Sunitinibin käytön yhteydessä on ilmoitettu verenpaineen kohoamista, myös vaikeaa verenpainetta (systolinen > 200 mmHg tai diastolinen 110 mmHg). Potilaat on tutkittava kohonneen verenpaineen varalta ja hoidettava asianmukaisesti. Hoidon tilapäistä keskeyttämistä suositellaan, jos potilaan vaikeasti kohonnutta verenpainetta ei saada hoidolla hallintaan. Sunitinibihoitoa voidaan jatkaa, kun kohonnut verenpaine on saatu asianmukaisesti hallintaan (ks. kohta Haittavaikutukset).
Hematologiset häiriöt
Sunitinibin käytön yhteydessä raportoitiin absoluuttisen neutrofiilimäärän ja verihiutalemäärän pienentymistä (ks. kohta Haittavaikutukset). Nämä tapahtumat eivät olleet kumulatiivisia vaan tyypillisesti korjaantuivat eivätkä yleensä johtaneet hoidon lopettamiseen. Yksikään kolmannen vaiheen tutkimuksissa todetuista tapahtumista ei ollut kuolemaan johtava, mutta valmisteen markkinoille tulon jälkeisessä seurannassa on joissakin harvinaisissa tapauksissa ilmoitettu hematologisia tapahtumia, mukaan lukien trombosytopeniaan ja neutropeenisiin infektioihin liittyviä verenvuotoja, jotka johtivat kuolemaan.
Anemiaa on havaittu ilmaantuvan sunitinibihoidon alkuvaiheessa sekä myöhemmin hoidon aikana.
Sunitinibihoitoa saavalta potilaalta tulee määrittää täydellinen verenkuva jokaisen hoitosyklin alussa (ks. kohta Haittavaikutukset).
Sydänhäiriöt
Sunitinibilla hoidetuilla potilailla on ilmoitettu sydän- ja verisuonitapahtumia, mukaan lukien sydämen vajaatoiminta, kardiomyopatia, vasemman kammion ejektiofraktion pieneneminen alle normaalin alaraja-arvon, sydänlihastulehdus, sydänlihasiskemia ja sydäninfarkti. Osa näistä tapahtumista on johtanut kuolemaan. Nämä tiedot viittaavat siihen, että sunitinibi lisää kardiomyopatian riskiä. Hoitoa saaneilla potilailla ei ole tunnistettu lääkeaineen spesifisen vaikutuksen lisäksi muita erityisiä riskitekijöitä sunitinibin aiheuttamaan kardiomyopatiaan. Sunitinibia on käytettävä varoen potilailla, joilla on riski saada tai joilla on aikaisemmin ilmennyt edellä mainittuja tapahtumia (ks. kohta Haittavaikutukset).
Kaikista sunitinibin kliinisistä tutkimuksista poissuljettiin potilaat, joilla oli ollut sunitinibin antoa edeltäneiden 12 kuukauden aikana jokin sydäntapahtuma, kuten sydäninfarkti (myös vaikea/epästabiili angina), sepelvaltimon/ääreisvaltimon ohitusleikkaus, oireinen kongestiivinen sydämen vajaatoiminta, aivoverisuonitapahtuma tai ohimenevä iskeeminen kohtaus (TIA) tai keuhkoveritulppa. Ei tiedetä, liittyykö sunitinibin käyttöön suurempi vasemman kammion toimintahäiriön kehittymisen riski potilailla, joilla on jokin edellä mainittu sairaus samanaikaisesti.
Lääkärin tulee arvioida riskiä lääkkeen mahdollisia hyötyjä vasten. Potilaita on seurattava sunitinibihoidon aikana tarkoin kongestiivisen sydämen vajaatoiminnan kliinisten merkkien ja oireiden varalta, erityisesti jos potilaalla on sydämeen liittyviä riskitekijöitä ja/tai hänellä on aikaisemmin todettu sepelvaltimotauti. Sydämen vasemman kammion ejektiofraktion mittaamista sekä lähtötilanteessa että ajoittain sunitinibihoidon aikana on harkittava. Ejektiofraktion mittaamista lähtötilanteessa on harkittava myös silloin, kun potilaalla ei ole sydämeen liittyviä riskitekijöitä.
Sunitinibihoidon lopettamista suositellaan, jos potilaalla ilmenee kliinisiä merkkejä kongestiivisesta sydämen vajaatoiminnasta. Sunitinibihoito on keskeytettävä ja/tai annosta pienennettävä potilaalla, jolla ei ole kliinistä näyttöä kongestiivisesta sydämen vajaatoiminnasta, mutta jolla ejektiofraktio on pienempi kuin 50 % ja laskee yli 20 % verrattuna lähtötasoon.
QT-ajan piteneminen
Sunitinibille altistuneilla potilailla on todettu QT-ajan pitenemistä ja kääntyvien kärkien takykardiaa (torsades de pointes). QT-ajan piteneminen voi suurentaa potilaan riskiä saada ventrikulaarisia rytmihäiriöitä, mukaan lukien kääntyvien kärkien takykardia.
Varovaisuutta on noudatettava sunitinibin käytössä potilaille, joiden QT-ajan tiedetään olleen aiemmin pidentynyt; potilaille, jotka käyttävät sydämen rytmihäiriölääkkeitä tai QT-aikaa mahdollisesti pidentäviä lääkevalmisteita; tai potilaille, joilla on entuudestaan jokin relevantti sydänsairaus, bradykardia tai elektrolyyttihäiriöitä. Sunitinibin samanaikaista antoa potenttien CYP3A4:n estäjien kanssa on vältettävä, koska plasman sunitinibipitoisuus voi suurentua (ks. kohdat Annostus ja antotapa, Yhteisvaikutukset ja Haittavaikutukset).
Laskimoiden tromboemboliset tapahtumat
Sunitinibia saaneilla potilailla on raportoitu hoitoon liittyneitä laskimoiden tromboembolisia tapahtumia, mukaan lukien syvä laskimotukos ja keuhkoveritulppa (ks. kohta Haittavaikutukset). Valmisteen markkinoille tulon jälkeisessä seurannassa on todettu kuolemaan johtaneita keuhkoveritulppatapauksia.
Valtimoiden tromboemboliset tapahtumat
Sunitinibihoitoa saaneilla potilailla on raportoitu valtimoiden tromboembolisia tapahtumia, jotka johtivat joskus potilaan kuolemaan. Yleisimpiä tapahtumia olivat aivoverisuonitapahtumat, ohimenevät aivojen verenkiertohäiriöt ja aivoinfarktit. Valtimoiden tromboembolisten tapahtumien riskitekijöitä olivat syöpäsairauden ja vähintään 65 vuoden iän lisäksi kohonnut verenpaine, diabetes ja aiempi tromboembolinen sairaus.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen sunitinibihoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Tromboottinen mikroangiopatia (TMA)
Tromboottisen mikroangiopatian diagnoosi, mukaan lukien tromboottinen trombosytopeeninen purppura (TTP) ja hemolyyttis-ureeminen oireyhtymä (HUS), on otettava huomioon, jos potilaalla ilmenee hemolyyttistä anemiaa, trombosytopeniaa, väsymystä, fluktuoivia neurologisia oireita ja löydöksiä, munuaisten vajaatoimintaa ja kuumetta. Tapaukset ovat joskus johtaneet munuaisten vajaatoimintaan tai kuolemaan. Sunitinibihoito tulisi lopettaa, jos potilaalle kehittyy tromboottinen mikroangiopatia. TMA:n hoito on aloitettava viipymättä. Tromboottisen mikroangiopatian oireiden häviämistä on todettu sunitinibihoidon keskeyttämisen jälkeen (ks. kohta Haittavaikutukset).
Kilpirauhasen toimintahäiriö
Jokaisen potilaan kilpirauhasen toiminta on suositeltavaa tutkia lähtötilanteessa laboratoriokokein. Kilpirauhasen vajaatoimintaa tai liikatoimintaa sairastavat potilaat tulee hoitaa tavanomaisen lääketieteellisen käytännön mukaisesti ennen sunitinibihoidon aloittamista. Sunitinibihoidon aikana kilpirauhasen toimintaa on seurattava säännöllisesti 3 kuukauden välein. Lisäksi potilaita on tarkkailtava tiiviisti kilpirauhasen toimintahäiriön merkkien ja oireiden varalta hoidon aikana. Jos potilaalle kehittyy kilpirauhasen toimintahäiriöön viittaavia merkkejä ja/tai oireita, kilpirauhasen toiminta on tutkittava laboratoriokokein kliinisen tarpeen mukaan. Potilaat, joille kehittyy kilpirauhasen toimintahäiriö, on hoidettava tavanomaisen lääketieteellisen käytännön mukaan.
Kilpirauhasen vajaatoimintaa on havaittu ilmaantuvan sunitinibihoidon alkuvaiheessa sekä myöhemmin hoidon aikana (ks. kohta Haittavaikutukset).
Haimatulehdus
Seerumin lipaasi- ja amylaasiaktiivisuuden lisääntymistä havaittiin sunitinibia saaneilla potilailla, joilla oli erilaisia kiinteitä kasvaimia. Näillä potilailla lipaasiaktiivisuuden lisääntyminen oli ohimenevää eikä siihen yleensä liittynyt haimatulehduksen merkkejä tai oireita (ks. kohta Haittavaikutukset).
Vakavia haimatapahtumia, joista osa on johtanut kuolemaan, on ilmoitettu. Jos potilaalla on haimatulehduksen oireita, sunitinibihoito on lopetettava ja potilaalle on annettava asianmukaista elintoimintoja tukevaa hoitoa.
Maksatoksisuus
Sunitinibihoitoa saaneilla potilailla on havaittu maksatoksisuutta. Maksan vajaatoimintaa, joka osassa tapauksista johti potilaan kuolemaan, havaittiin alle 1 %:lla sunitinibihoitoa saaneista potilaista, joilla oli kiinteä kasvain. Maksan toimintakokeiden tuloksia (alaniinitransaminaasi- [ALAT], aspartaattitransaminaasi- [ASAT], bilirubiinipitoisuuksia) on seurattava ennen hoidon aloittamista, kunkin hoitosyklin aikana ja kliinisen tarpeen mukaan. Jos maksan vajaatoiminnan merkkejä tai oireita ilmenee, sunitinibihoito on lopetettava ja potilaalle on annettava asianmukaista elintoimintoja tukevaa hoitoa (ks. kohta Haittavaikutukset).
Munuaisten toiminta
Munuaisten toimintahäiriöitä, vajaatoimintaa ja/tai akuuttia vajaatoimintaa on raportoitu ja ne ovat johtaneet joissakin tapauksissa potilaan kuolemaan (ks. kohta Haittavaikutukset).
Taustalla olevan munuaissolukarsinooman lisäksi munuaisten toimintahäiriön/vajaatoiminnan riskitekijöitä sunitinibia saavilla potilailla ovat korkea ikä, diabetes, taustalla oleva munuaisten toimintahäiriö, sydämen vajaatoiminta, kohonnut verenpaine, sepsis, dehydraatio/hypovolemia ja rabdomyolyysi.
Sunitinibihoidon jatkamisen turvallisuutta potilailla, joilla on keskivaikea tai vaikea proteinuria, ei ole arvioitu järjestelmällisesti.
Proteinuriaa ja joissakin harvinaisissa tapauksissa nefroottista oireyhtymää on ilmoitettu. Virtsakokeiden tekemistä lähtötilanteessa suositellaan, ja potilasta tulee seurata proteinurian kehittymisen tai pahenemisen varalta. Sunitinibihoito on lopetettava, jos potilaalla ilmenee nefroottinen oireyhtymä.
Fistelit
Jos fistelimuodostusta ilmenee, sunitinibihoito on keskeytettävä. Sunitinibihoidon jatkamisesta potilaille, joilla on havaittu fisteleitä, on saatavilla vain vähän tietoa (ks. kohta Haittavaikutukset).
Heikentynyt haavojen paraneminen
Haavojen paranemisen on raportoitu heikentyneen sunitinibihoidon aikana.
Sunitinibin vaikutusta haavojen paranemiseen ei ole tutkittu systemaattisissa kliinisissä tutkimuksissa. Jos potilaalle suunnitellaan suurta leikkausta, sunitinibihoito suositellaan varotoimena keskeyttämään tilapäisesti. Hoidon uudelleen aloittamisen ajankohdasta suuren leikkauksen jälkeen on vähän kliinistä kokemusta. Päätöksen sunitinibihoidon aloittamisesta uudelleen suuren leikkauksen jälkeen on siksi perustuttava kliiniseen arvioon siitä, miten potilas on toipunut leikkauksesta.
Leukaluun osteonekroosi
Sunitinibi-valmisteella hoidetuilla potilailla on raportoitu leukaluun osteonekroosia. Suurin osa tapauksista raportoitiin potilailla, jotka olivat saaneet edeltävää tai samanaikaista laskimonsisäistä bisfosfonaattilääkitystä. Bisfosfonaattien laskimonsisäiseen käyttöön tiedetään liittyvän leukaluun osteonekroosin riski. Varovaisuutta on siksi noudatettava, kun Sunitinib Krka -valmistetta ja laskimonsisäisiä bisfosfonaatteja käytetään joko samanaikaisesti tai peräkkäin.
Invasiiviset hammastoimenpiteet on myös tunnistettu riskitekijäksi. Ennen Sunitinib Krka -hoidon aloittamista on harkittava hampaiden kunnon tarkistamista ja sopivaa ehkäisevää hammashoitoa. Invasiivisia hammastoimenpiteitä on mahdollisuuksien mukaan vältettävä, jos potilas on saanut aiemmin tai saa parhaillaan bisfosfonaatteja laskimonsisäisesti (ks. kohta Haittavaikutukset).
Yliherkkyys/angioödeema
Jos yliherkkyyden aiheuttamaa angioödeemaa ilmenee, sunitinibihoito on keskeytettävä ja potilasta on hoidettava asianmukaisesti (ks. kohta Haittavaikutukset).
Kouristuskohtaukset
Sunitinibilla tehdyissä kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeisessä seurannassa on raportoitu kouristuskohtauksia. Jos potilaalla ilmenee kouristuskohtauksia ja merkkejä/oireita, jotka viittaavat korjaantuvaan posterioriseen leukoenkefalopatiaoireyhtymään (esim. kohonnut verenpaine, päänsärky, vireystason heikkeneminen, mentaalisten toimintojen muuttuminen ja näönmenetys, mukaan lukien kortikaalinen sokeus), oireet, myös kohonnut verenpaine, on saatava hallintaan. Sunitinibihoidon tilapäistä keskeyttämistä suositellaan: kun oireet häviävät, hoito voidaan aloittaa uudelleen hoitavan lääkärin harkinnan mukaan (ks. kohta Haittavaikutukset).
Tuumorilyysioireyhtymä (TLS)
Tuumorilyysioireyhtymätapauksia, joista osa johti kuolemaan, on todettu harvoin sunitinibihoitoa saaneilla potilailla kliinisissä tutkimuksissa ja markkinoille tulon jälkeisissä seurannassa. Tuumorilyysioireyhtymän riskitekijöitä ovat suuri tuumoritaakka, krooninen munuaisten vajaatoiminta, oliguria, dehydraatio, hypotensio ja virtsan happamuus. Näitä potilaita on tarkkailtava huolellisesti ja hoidettava tarpeen mukaan. Profylaktista nesteytystä on harkittava.
Infektiot
Vakavia infektioita, joissakin tapauksissa kuolemaan johtaneita, on raportoitu neutropenian yhteydessä ja ilman neutropeniaa. Nekrotisoivaa faskiittia, myös välilihassa, on raportoitu melko harvoin. Osa tapauksista on johtanut kuolemaan (ks. kohta Haittavaikutukset).
Jos potilaalle kehittyy nekrotisoiva faskiitti, sunitinibihoito tulee keskeyttää ja aloittaa viipymättä tilanteen edellyttämä hoito.
Hypoglykemia
Sunitinibihoidon aikana on ilmoitettu verensokerin laskua, joka on joissakin tapauksissa aiheuttanut kliinisiä oireita ja vaatinut sairaalahoitoa tajunnan menetyksen vuoksi. Sunitinibihoito tulee keskeyttää tilapäisesti, jos oireista hypoglykemiaa ilmenee. Diabeetikkojen verensokeriarvo tulee tarkistaa säännöllisesti, jotta diabeteslääkevalmisteen annosta voidaan tarvittaessa muuttaa hypoglykemiariskin minimoimiseksi (ks. kohta Haittavaikutukset).
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kapseli eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia on tehty vain aikuisilla.
Lääkevalmisteet, jotka voivat suurentaa plasman sunitinibipitoisuuksia
CYP3A4:n estäjien vaikutus
Kun terveille vapaaehtoisille tutkimushenkilöille annettiin kerta-annos sunitinibimalaattia samanaikaisesti vahvan CYP3A4:n estäjän ketokonatsolin kanssa, sunitinibin ja päämetaboliitin yhdistetty enimmäispitoisuus (Cmax) suureni 49 % ja pitoisuus-aikakuvaajan alle jäävä pinta-ala (AUC0-∞) 51 %.
Sunitinibin anto yhdessä vahvojen CYP3A4:n estäjien (kuten ritonaviirin, itrakonatsolin, erytromysiinin, klaritromysiinin, greippimehun) kanssa voi suurentaa sunitinibin pitoisuuksia.
Siksi samanaikaista hoitoa CYP3A4:n estäjien kanssa on vältettävä tai valittava muu samanaikainen vaihtoehtoinen lääkevalmiste, joka ei estä CYP3A4:ää lainkaan tai mahdollisesti vain vähän.
Jos tämä ei ole mahdollista, Sunitinib Krka -annos on ehkä pienennettävä vähimmäismäärään 37,5 mg/vrk GIST- ja MRCC-käyttöaiheissa tai vähimmäismäärään 25 mg/vrk haiman NET -käyttöaiheessa siedettävyyden huolellisen seurannan perusteella (ks. kohta Annostus ja antotapa).
Rintasyövän resistenssiproteiinin (BCRP:n) estäjien vaikutus
Saatavilla on vain vähän kliinisiä tietoja sunitinibin ja BCRP:n estäjien välisestä yhteisvaikutuksesta, eikä yhteisvaikutuksen mahdollisuutta sunitinibin ja muiden BCRP:n estäjien välillä voida sulkea pois (ks. kohta Farmakokinetiikka).
Lääkevalmisteet, jotka voivat pienentää plasman sunitinibipitoisuuksia
CYP3A4:n induktorien vaikutus
Kun terveille vapaaehtoisille tutkimushenkilöille annettiin kerta-annos sunitinibia samanaikaisesti CYP3A4:n induktorin rifampisiinin kanssa, sunitinibin ja päämetaboliitin yhdistetty Cmax-arvo pieneni 23 % ja AUC0-∞-arvo 46 %.
Sunitinibin anto yhdessä vahvojen CYP3A4:n induktorien (kuten deksametasonin, fenytoiinin, karbamatsepiinin, rifampisiinin, fenobarbitaalin tai mäkikuismaa eli Hypericum perforatumia sisältävien rohdosvalmisteiden) kanssa voi pienentää sunitinibin pitoisuuksia. Siksi samanaikaista hoitoa CYP3A4:n induktorien kanssa on vältettävä tai valittava muu samanaikainen vaihtoehtoinen lääkevalmiste, joka ei indusoi CYP3A4:ää lainkaan tai mahdollisesti vain vähän. Jos tämä ei ole mahdollista, Sunitinib Krka -annostusta on ehkä suurennettava 12,5 mg/vrk:n kerta-annoksin (enintään määrään 87,5 mg/vrk GIST- ja MRCC-käyttöaiheissa tai määrään 62,5 mg/vrk haiman NET -käyttöaiheessa) siedettävyyden huolellisen seurannan perusteella (ks. kohta Annostus ja antotapa).
Raskaus ja imetys
Ehkäisy miehillä ja naisilla
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä ja välttämään raskaaksituloa sunitinibihoidon aikana.
Raskaus
Sunitinibin käyttöä raskaana oleville naisille ei ole tutkittu. Eläintutkimuksissa on osoitettu lisääntymistoksisuutta, myös sikiön epämuodostumia (ks. kohta Prekliiniset tiedot turvallisuudesta). Sunitinibia ei saa käyttää raskausaikana eikä naispotilaille, jotka eivät käytä tehokasta raskaudenehkäisyä, jollei mahdollinen hyöty oikeuta sikiölle koituvaa mahdollista riskiä. Jos sunitinibia annetaan raskaana olevalle tai jos potilas tulee raskaaksi sunitinibihoidon aikana, hänelle on kerrottava sikiölle mahdollisesti aiheutuvasta vaarasta.
Imetys
Sunitinibi ja/tai sen metaboliitit erittyvät rotan rintamaitoon. Ei tiedetä, erittyykö sunitinibi tai sen aktiivinen päämetaboliitti ihmisen rintamaitoon. Koska vaikuttavat aineet yleensä erittyvät ihmisen rintamaitoon ja koska rintaruokitut lapset voivat saada tästä vakavia haittavaikutuksia, sunitinibihoidon aikana ei saa imettää.
Hedelmällisyys
Ei-kliinisten tutkimusten tulosten perusteella sunitinibihoito voi vaarantaa sekä miehen että naisen hedelmällisyyden (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Sunitinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilasta on varoitettava, että hänellä voi ilmetä heitehuimausta sunitinibihoidon aikana.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Vakavimmat sunitinibiin liittyvät haittavaikutukset ovat munuaisten vajaatoiminta, sydämen vajaatoiminta, keuhkoveritulppa, ruoansulatuskanavan puhkeaminen ja verenvuodot (esim. hengitysteissä, ruoansulatuskanavassa, kasvaimessa, virtsateissä ja aivoissa). Osa tapauksista johti kuolemaan. Yleisimpiä kaikenasteisia haittavaikutuksia (ilmeni RCC-, GIST- ja haiman NET- rekisteröintitutkimuksissa mukana olleilla potilailla) olivat ruokahalun väheneminen, makuaistin häiriöt, kohonnut verenpaine, uupumus, ruoansulatuskavanan häiriöt (esim. ripuli, pahoinvointi, suutulehdus, dyspepsia ja oksentelu), ihon värjäytyminen sekä käsi-jalkaoireyhtymä. Nämä oireet saattavat vähentyä hoitoa jatkettaessa. Kilpirauhasen vajaatoiminta saattaa kehittyä hoidon aikana. Hematologiset häiriöt (kuten neutropenia, trombosytopenia ja anemia) kuuluvat yleisimpien haittavaikutusten joukkoon.
Edellä kohdassa Varoitukset ja käyttöön liittyvät varotoimet ja tässä kohdassa Haittavaikutukset jäljempänä lueteltujen kuolemaan johtaneiden tapahtumien lisäksi muita sunitinibiin mahdollisesti liittyviä kuolemaan johtaneita tapahtumia olivat monielinhäiriö, disseminoitunut intravaskulaarinen koagulaatio, peritoneaalinen verenvuoto, lisämunuaisten vajaatoiminta, ilmarinta, sokki ja äkkikuolema.
Taulukoitu luettelo haittavaikutuksista
Haittavaikutukset, joita raportoitiin GIST-, MRCC- ja haiman NET -potilailla, kun 7115 potilaasta kerätyt tiedot yhdistettiin, on lueteltu alla elinjärjestelmän, esiintymistiheyden ja vaikeusasteen mukaan (NCI-CTCAE). Mukana ovat myös haittavaikutukset, jotka on todettu markkinoille tulon jälkeen kliinisissä tutkimuksissa. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan laskevassa järjestyksessä.
Esiintymistiheyksien määritelmät: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 1. Kliinisissä tutkimuksissa ilmoitetut haittavaikutukset
Elinjärjestel-mä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
Infektiot | Virusinfektiota Hengitystieinfektiot b,* Absessi c,* Sieni-infektiotd Virtsatieinfektio Ihoinfektiote Sepsis f,* | Nekrotisoiva faskiitti* Bakteeriinfektiotg | |||
Veri ja imukudos | Neutropenia Trombosytopenia Anemia Leukopenia | Lymfosytopenia | Pansytopenia | Tromboottinen mikroangiopatia h,* | |
Immuuni- järjestelmä | Yliherkkyys | Angioedeema | |||
Umpieritys | Kilpirauhasen vajaatoiminta | Kilpirauhasen liikatoiminta | Kilpirauhastu-lehdus | ||
Aineenvaih- dunta ja ravitsemus | Ruokahalun vähenemineni | Elimistön kuivuminen Hypoglykemia | Tuumorilyysi- oireyhtymä* | ||
Psyykkiset häiriöt | Unettomuus | Masennus | |||
Elinjärjestel-mä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
Hermosto | Heitehuimaus Päänsärky Makuaistin häiriöj | Perifeerinen neuropatia Parestesia Hypoestesia Hyperestesia | Aivoverenvuoto* Aivoverisuoni-tapahtuma* Ohimenevä iskeeminen kohtaus | Posteriorinen reversiibeli enkefalopatia-oireyhtymä* | |
Silmät | Periorbitaalinen edeema Silmäluomien turvotus Kyynelnesteen lisääntyminen | ||||
Sydän | Sydänlihasiskemia k,* Ejektiofraktion laskul | Kongestiivinen sydämen vajaatoiminta Sydäninfarkti m,* Sydämen vajaatoiminta* Kardiomyopatia* Perikardiaalinen effuusio Pidetynyt QT-aika EKG:ssä | Vasemman kammion vajaatoiminta* Kääntyvien kärkien takykardia (torsades de pointes) | ||
Verisuonisto | Kohonnut verenpaine | Syvä laskimotromboosi Kuumat aallot Kasvojen punoitus ja kuumotus | Verenvuoto kasvaimessa* | Aneurysmat ja valtimon dissekaatiot* | |
Hengityselimet, rintakehä ja välikarsina | Hengenahdistus Nenäverenvuoto Yskä | Keuhkoembolia* Pleuraeffuusio* Veriyskös Rasitukseen liittyvä hengenahdistus Suunielun kipun Nenän tukkoisuus Nenän kuivuminen | Keuhkoveren-vuoto* Hengityksen vajaatoiminta* | ||
Ruoansula-tuselimistö | Stomatiittio Vatsakipup Oksentelu Ripuli Dyspepsia Pahoinvointi Ummetus | Gastroesofageaalinen refluksitauti Dysfagia Ruoansulatuskanavan verenvuoto* Esofagiitti* Vatsan pingotus Vatsavaivat Verenvuoto peräsuolesta Verenvuoto ikenistä Suun haavautuminen Peräsuolikipu Huulitulehdus Peräpukamat Kielikipu Suukipu Suun kuivuminen Ilmavaivat Epämiellyttävä tunne suussa Röyhtäily | Ruoansulatus- kanavan puhkeaminen q,* Haimatulehdus Peräaukon fisteli Koliittir | ||
Elinjärjestel-mä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
Maksa ja sappi | Maksan vajaatoiminta* Sappirakkotuleh-dus s,* Maksan toimintahäiriö | Hepatiitti | |||
Iho ja ihonalainen kudos | Ihon värimuutoksett Käsi-jalka- oireyhtymä Ihottumau Hiusten värimuutokset Ihon kuivuminen | Ihon hilseily Ihoreaktiov Ekseema Rakkula Punoitus Hiustenlähtö Akne Kutina Ihon hyperpigmentaatio Iholeesio Hyperkeratoosi Ihotulehdus Kynsihäiriöw | Erythema multiforme* Stevens-Johnsonin oireyhtymä* Pyoderma gangrenosum Toksinen epidermaalinen nekrolyysi* | ||
Luusto, lihakset ja sidekudos | Raajakipu Nivelkipu Selkäkipu | Tuki- ja liikuntaelimistön kipu Lihasspasmit Lihaskipu Lihasheikkous | Leukaluun osteonekroosi Fisteli* | Rabdomyolyysi* Myopatia | |
Munuaiset ja virtsatiet | Munuaisten vajaatoiminta* Akuutti munuaisten vajaatoiminta* Kromaturia Proteinuria | Verenvuoto virtsateissä | Nefroottinen oireyhtymä | ||
Yleisoireet ja antopaikassa todettavat haitat | Limakalvotuleh-dus Väsymysx Edeemay Kuume | Rintakipu Kipu Influenssan kaltainen oireisto Vilunväristykset | Heikentynyt haavojen paraneminen | ||
Tutkimukset | Painon lasku Valkosolumäärän pieneneminen Lipaasiarvon suureneminen Verihiutalemäärän pieneneminen Hemoglobiiniarvon pieneneminen Amylaasiarvon suureneminenz Aspartaattiamino-transferaasiarvon suureneminen Alaniiniamino-transferaasiarvon suureneminen Kreatiniiniarvon suureneminen Verenpaineen nousu Virtsahappopitoisuu-den suureneminen veressä | Veren kreatiinikinaasiarvon suureneminen Veren tyreotropiinin nousu |
* mukaan lukien kuolemaan johtaneet tapaukset
Seuraavat termit on yhdistetty:
a nasofaryngiitti ja suuherpes
b keuhkoputkitulehdus, alahengitystieinfektio, keuhkokuume ja hengitystieinfektio
c paise, paise raajassa, peräaukon paise, ikenen märkäpesäke, maksapaise, haiman märkäpesäke, välilihan paise, peräsuolenvieruspaise, peräsuolipaise, ihonalainen paise ja hammasabsessi
d ruokatorven kandidiaasi ja suun kandidiaasi
e selluliitti ja ihoinfektio
f sepsis ja septinen sokki
g vatsaontelon sisäinen märkäkertymä, vatsaontelon sepsis, divertikuliitti ja osteomyeliitti
h tromboottinen mikroangiopatia, tromboottinen trombosytopeeninen purppura, hemolyyttis-ureeminen oireyhtymä
i vähentynyt ruokahalu ja ruokahaluttomuus
j dysgeusia, ageusia ja makuaistin häiriö
k akuutti koronaarisyndrooma, angina pectoris, epästabiili angina, sepelvaltimotukos, sydänlihasiskemia
l ejektiofraktion lasku/poikkeama
m akuutti sydäninfarkti, sydäninfarkti, oireeton sydäninfarkti
n suunielun sekä nielun ja kurkunpään kipu
o stomatiitti ja aftainen stomatiitti
p vatsakipu, alavatsan ja ylävatsan kipu
q ruoansulatuskanavan puhkeaminen ja suoliston puhkeaminen
r koliitti ja iskeeminen koliitti
s sappirakkotulehdus ja kivetön sappirakkotulehdus
t ihon kellertäminen, ihon värjäytyminen ja pigmenttihäiriö
u psoriasiforminen dermatiitti, kesivä ihottuma, ihottuma, punoittava ihottuma, follikulaarinen ihottuma, yleistynyt ihottuma, täpläinen ihottuma, täpläinen ja näppyläinen ihottuma, näppyläinen ihottuma ja kutiava ihottuma
v ihoreaktio ja ihosairaus
w kynsihäiriö ja kynsien värjäytyminen
x väsymys ja astenia
y kasvojen edeema, edeema ja perifeerinen edeema
z amylaasiarvo ja amylaasiarvon suureneminen
Valittujen haittavaikutusten kuvaus
Infektiot
Vakavia infektioita (joihin liittyi neutropenia tai joihin sitä ei liittynyt), mukaan lukien kuolemaan johtaneita tapauksia, on raportoitu. Nekrotisoivaa faskiittia, myös välilihassa, on raportoitu. Osa tapauksista on johtanut kuolemaan (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Veri ja imukudos
Absoluuttisen neutrofiilimäärän pienentymistä vaikeusasteelle 3 ilmoitettiin 10 %:lla potilaista ja vaikeusasteelle 4 1,7 %:lla potilaista kolmannen vaiheen GIST-tutkimuksessa. Vastaavat luvut kolmannen vaiheen MRCC-tutkimuksessa olivat 16 % ja 1,6 % ja kolmannen vaiheen haiman NET - tutkimuksessa 13 % ja 2,4 %. Verihiutalemäärän pienentymistä vaikeusasteelle 3 ilmoitettiin 3,7 %:lla potilaista ja vaikeusasteelle 4 0,4 %:lla potilaista kolmannen vaiheen GIST-tutkimuksessa. Vastaavat luvut kolmannen vaiheen MRCC-tutkimuksessa olivat 8,2 % ja 1,1 % ja kolmannen vaiheen haiman NET -tutkimuksessa 3,7 % ja 1,2 % (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenvuototapahtumia ilmeni 18 %:lla sunitinibia saaneista potilaista ja 17 %:lla lumelääkettä saaneista potilaista kolmannen vaiheen GIST-tutkimuksessa. Aiemmin hoitamattomassa MRCC:ssa verenvuototapahtumia ilmeni 39 %:lla sunitinibia saaneista potilaista ja 11 %:lla α- interferonipotilaista. Vähintään vaikeusasteen 3 verenvuototapahtumia ilmeni 17 sunitinibipotilaalla (4,5 %) ja viidellä α-interferonipotilaalla (1,7 %). Niistä potilaista, jotka saivat sunitinibia sytokiini-refraktaariseen MRCC:hen, verenvuotoa ilmeni 26 %:lla. Kolmannen vaiheen haiman NET -tutkimuksessa verenvuototapahtumia (pois lukien nenäverenvuoto) raportoitiin 21,7 %:lla potilaista sunitinibiryhmässä ja 9,85 %:lla lumelääkeryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisissä tutkimuksissa kasvaimen verenvuotoa raportoitiin noin 2 %:lla GIST-potilaista.
Immuunijärjestelmä
Yliherkkyysreaktioita, mukaan lukien angioedeemaa, on raportoitu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Umpieritys
Kahdessa sytokiini-refraktaarista MRCC:tä koskeneessa tutkimuksessa ilmoitettiin haittavaikutuksena kilpirauhasen vajaatoiminta seitsemällä sunitinibia saaneella potilaalla (4 %). Tutkimuksessa, joka koski aiemmin hoitamatonta MRCC:tä, ilmoitettiin kilpirauhasen vajaatoimintaa 61 potilaalla (16 %) sunitinibiryhmässä ja 3 potilaalla (< 1 %) α-interferoniryhmässä.
Lisäksi neljällä sytokiini-refraktaarisella MRCC-potilaalla (2 %) ilmoitettiin tyreotropiinin (TSH) nousua. Kaiken kaikkiaan 7 %:lla MRCC-tutkimuspopulaatiosta ilmeni joko kliinistä tai laboratoriotutkimuksiin perustuvaa näyttöä hoidon aikana alkaneesta kilpirauhasen vajaatoiminnasta. Hankittu kilpirauhasen vajaatoiminta ilmeni 6,2 %:lla sunitinibia saaneista ja 1 %:lla lumelääkettä saaneista GIST-potilaista. Kolmannen vaiheen haiman NET -tutkimuksessa kilpirauhasen vajaatoimintaa raportoitiin kuudella sunitinibia saaneella potilaalla (7,2 %) ja yhdellä lumelääkettä saaneella potilaalla (1,2 %).
Kilpirauhasen toimintaa seurattiin prospektiivisesti kahdessa rintasyöpäpotilailla tehdyssä tutkimuksessa. Sunitinibia ei ole hyväksytty rintasyövän hoitoon. Yhdessä tutkimuksessa kilpirauhasen vajaatoimintaa raportoitiin 15:lla sunitinibia saaneella (13,6 %) ja 3:lla tavanomaista hoitoa saaneella potilaalla (2,9 %). Veren tyreotropiinin nousua raportoitiin yhdellä sunitinibia saaneella potilaalla (0,9 %), mutta ei yhdelläkään tavanomaista hoitoa saaneista. Kilpirauhasen liikatoimintaa ei raportoitu sunitinibia saaneilla potilailla, mutta sitä raportoitiin yhdellä tavanomaista hoitoa saaneista (1,0 %). Toisessa tutkimuksessa kilpirauhasen vajaatoimintaa raportoitiin yhteensä 31:llä sunitinibia saaneella potilaalla (13 %) ja kahdella kapesitabiinia saaneista potilaista (0,8 %). Veren tyreotropiinin nousua raportoitiin 12:lla sunitinibia saaneella potilaalla (5,0 %), mutta ei yhdelläkään kapesitabiinia saaneista. Kilpirauhasen liikatoimintaa raportoitiin neljällä sunitinibia saaneella potilaalla (1,7 %), mutta ei yhdelläkään kapesitabiinia saaneista. Veren tyreotropiinin laskua raportoitiin kolmella sunitinibia saaneella potilaalla (1,3 %), mutta ei yhdelläkään kapesitabiinia saaneista. Kilpirauhashormonin (T4) nousua raportoitiin kahdella sunitinibia saaneella (0,8 %) ja yhdellä kapesitabiinia saaneella potilaalla (0,4 %). Trijodotyroniinin (T3) nousua raportoitiin yhdellä sunitinibia saaneella potilaalla (0,8 %), mutta ei yhdelläkään kapesitabiinia saaneista. Kaikki raportoidut kilpirauhasen toimintaan liittyneet tapahtumat olivat vaikeusastetta 1 tai 2 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aineenvaihdunta ja ravitsemus
Haiman NET -potilailla raportoitiin hypoglykemiatapauksia suuremmalla esiintymistiheydellä kuin MRCC- ja GIST-potilailla. Suurinta osaa näistä kliinisissä tutkimuksissa havaituista haittatapahtumista ei kuitenkaan pidetty tutkimushoitoon liittyvinä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hermosto
Sunitinibilla tehdyissä kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeisessä seurannassa on saatu harvoin (< 1 %) raportteja potilaista, joilla on ilmennyt kouristuskohtauksia ja joilla on radiologisesti todettu posteriorinen reversiibeli leukoenkefalopatiaoireyhtymä (RPLS). Osa tapauksista on johtanut kuolemaan. Kouristuskohtauksia on ilmennyt potilailla, joilla on radiologisesti todettu etäpesäkkeitä aivoissa, mutta myös potilailla ilman radiologista näyttöä aivojen etäpesäkkeistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydän
Kliinisissä tutkimuksissa vasemman kammion ejektiofraktion (LVEF-arvo) pienenemistä ≥ 20 % ja alle normaalin alaraja-arvon raportoitiin noin 2 %:lla sunitinibilla hoidetuista GIST-potilaista, 4 %:lla sytokiini-refraktaarisista MRCC-potilaista ja 2 %:lla lumelääkkeellä hoidetuista GIST-potilaista. LVEF-arvojen pieneneminen ei näytä olleen progressiivista vaan LVEF-arvo parani usein hoidon jatkuessa. Tutkimuksessa, joka koski aiemmin hoitamatonta MRCC:tä, LVEF-arvo oli alle normaalin alarajan 27 %:lla sunitinibipotilaista ja 15 %:lla α-interferonipotilaista. Kahdella sunitinibia saaneella potilaalla (< 1 %) diagnosoitiin kongestiivinen sydämen vajaatoiminta.
Sydämen vajaatoimintaa, kongestiivista sydämen vajaatoimintaa ja sydämen vasemman kammion vajaatoimintaa ilmoitettiin 1,2 %:lla sunitinibilla hoidetuista GIST-potilaista ja 1 %:lla lumelääkkeellä hoidetuista GIST-potilaista. Keskeisessä kolmannen vaiheen GIST-tutkimuksessa (N = 312) raportoitiin hoitoon liittyneitä kuolemaan johtaneita sydänhaittoja 1 %:lla potilaista kummassakin tutkimusryhmässä (eli sekä sunitinibi- että lumelääkeryhmässä). Toisen vaiheen sytokiini- refraktaarista MRCC:tä koskeneessa tutkimuksessa 0,9 %:lla potilaista ilmeni hoitoon liittynyt kuolemaan johtanut sydäninfarkti. Kolmannen vaiheen tutkimuksessa, johon osallistui aiemmin hoitoa saamattomia MRCC-potilaita, kuolemaan johtaneita sydäntapahtumia ilmeni 0,6 %:lla α- interferonipotilaista, mutta ei yhdelläkään (0 %) sunitinibipotilaalla. Kolmannen vaiheen haiman NET -tutkimuksessa yhdellä sunitinibia saaneella potilaalla (1 %) ilmeni hoitoon liittynyt kuolemaan johtanut sydämen vajaatoiminta.
Verisuonisto
Kohonnut verenpaine
Verenpaineen kohoaminen oli hyvin yleisesti raportoitu haittavaikutus kliinisissä tutkimuksissa. Sunitinibiannosta pienennettiin tai sen anto keskeytettiin tilapäisesti noin 2,7 %:lla potilaista, joilla todettiin verenpainetta. Yhdenkään potilaan sunitinibihoitoa ei lopetettu pysyvästi. Vaikeaa verenpaineen kohoamista (systolinen > 200 mmHg tai diastolinen 110 mmHg) raportoitiin 4,7 %:lla potilaista, joilla oli kiinteitä kasvaimia. Verenpaineen kohoamista ilmoitettiin noin 33,9 %:lla potilaista, jotka saivat sunitinibia aiemmin hoitamattomaan MRCC:hen, verrattuna 3,6 %:iin potilaista, jotka saivat α-interferonia. Vaikeaa verenpaineen kohoamista raportoitiin 12 %:lla aiemmin hoitamattomista sunitinibipotilaista ja < 1 %:lla α-interferonipotilaista. Kolmannen vaiheen haiman NET -tutkimuksessa kohonnutta verenpainetta raportoitiin 26,5 %:lla potilaista sunitinibiryhmässä ja 4,9 %:lla lumelääkeryhmässä. Vaikeaa verenpaineen kohoamista ilmeni 10 %:lla sunitinibia saaneista haiman NET -potilaista ja 3 %:lla lumelääkettä saaneista potilaista.
Laskimoiden tromboemboliset tapahtumat
Hoitoon liittyneitä laskimoiden tromboembolisia tapahtumia ilmoitettiin kliinisissä tutkimuksissa noin 1,0 %:lla sunitinibia saaneista potilaista, joilla oli kiinteitä kasvaimia, mukaan lukien GIST ja RCC.
Kolmannen vaiheen GIST-tutkimuksessa laskimoiden tromboembolisia tapahtumia ilmeni seitsemällä sunitinibia saaneella potilaalla (3 %), mutta ei yhdelläkään lumelääkettä saaneista: näistä seitsemästä potilaasta viidellä ilmeni vaikeusasteen 3 ja kahdella vaikeusasteen 1 tai 2 syvä laskimotukos. Näistä seitsemästä GIST-potilaasta neljällä hoito lopetettiin heti, kun syvä laskimotukos oli havaittu.
Kolmannen vaiheen tutkimuksessa, joka koski aiemmin hoitamatonta MRCC:tä, laskimoiden tromboembolisia tapahtumia ilmoitettiin kolmellatoista sunitinibia saaneella potilaalla (3 %) ja kahdessa sytokiini-refraktaarista MRCC:tä koskeneessa tutkimuksessa 4 potilaalla (2 %). Näistä potilaista yhdeksällä ilmeni keuhkoveritulppa: yksi tapaus oli vaikeusastetta 2 ja kahdeksan tapausta vaikeusastetta 4. Kahdeksalla näistä potilaista ilmeni syvä laskimotukos: yksi tapaus vaikeusastetta 1, kaksi tapausta vaikeusastetta 2, neljä tapausta vaikeusastetta 3 ja yksi tapaus vaikeusastetta 4. Sytokiini-refraktaarista MRCC:tä koskeneessa tutkimuksessa annostelu keskeytettiin yhdellä potilaalla, jolla esiintyi keuhkoveritulppa.
Aiemmin hoitamattomien MRCC-potilaiden α-interferoniryhmässä 6 potilaalla (2 %) ilmeni laskimoiden tromboembolisia tapahtumia: yhdellä potilaalla (< 1 %) ilmeni vaikeusasteen 3 syvä laskimotukos ja viidellä potilaalla (1 %) keuhkoveritulppa, kaikki vaikeusastetta 4.
Kolmannen vaiheen haiman NET -tutkimuksessa laskimoiden tromboembolisia tapahtumia raportoitiin yhdellä potilaalla (1,2 %) sunitinibihaarassa ja 5 potilaalla (6,1 %) lumelääkehaarassa. Kahdella lumelääkettä saaneella potilaalla oli syvä laskimotukos, toisella vaikeusastetta 2 ja toisella vaikeusastetta 3.
Yhtään kuolemantapausta ei ilmoitettu GIST-, MRCC- ja haiman NET -rekisteröintitutkimuksissa. Valmisteen markkinoille tulon jälkeisessä seurannassa on havaittu kuolemaan johtaneita tapauksia.
Kolmannen vaiheen tutkimuksissa keuhkoveritulppia ilmoitettiin noin 3,1 %:lla sunitinibia saaneista GIST-potilaista ja noin 1,2 %:lla sunitinibia saaneista MRCC-potilaista. Kolmannen vaiheen haiman NET -tutkimuksessa sunitinibia saaneilla potilailla ei raportoitu keuhkoveritulppia. Harvoja kuolemaan johtaneita tapauksia on havaittu markkinoille tulon jälkeisessä seurannassa.
Sunitinibin kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli ollut keuhkoveritulppa edeltäneiden 12 kuukauden aikana.
Kolmannen vaiheen rekisteröintitutkimuksissa keuhkotapahtumia (eli hengenahdistus, pleuraeffuusio, keuhkoveritulppa tai keuhkoedeema) ilmoitettiin noin 17,8 %:lla sunitinibia saaneista GIST-potilaista, noin 26,7 %:lla sunitinibia saaneista MRCC-potilaista ja 12 %:lla sunitinibia saaneista haiman NET - potilaista.
Kliinisissä tutkimuksissa sunitinibia saaneista potilaista, joilla oli kiinteä kasvain (mukaan lukien GIST ja MRCC), keuhkotapahtumia ilmoitettiin noin 22,2 %:lla.
Ruoansulatuselimistö
Haimatulehdusta on todettu melko harvoin (< 1 %) potilailla, jotka saavat sunitinibihoitoa GIST:iin tai MRCC:hen. Kolmannen vaiheen haiman NET -tutkimuksessa ei raportoitu hoitoon liittyvää haimatulehdusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kolmannen vaiheen GIST-tutkimuksessa ilmeni kuolemaan johtanut ruoansulatuskanavan verenvuoto 0,98 %:lla lumelääkettä saaneista potilaista.
Maksa ja sappi
Maksan toimintahäiriöitä on raportoitu. Näitä voivat olla maksan toimintakokeiden poikkeavuudet, hepatiitti tai maksan vajaatoiminta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iho ja ihonalainen kudos
Pyoderma gangrenosum -tapauksia on raportoitu. Nämä tapaukset yleensä paranivat, kun sunitinibin anto keskeytettiin (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Luusto, lihakset ja sidekudos
Joissakin tapauksissa on ilmoitettu myopatiaa ja/tai rabdomyolyysiä, joista osaan liittyi akuutti munuaisten vajaatoiminta. Potilasta, jolla ilmenee lihastoksisuuden merkkejä tai oireita, tulee hoitaa tavanomaisen lääketieteellisen käytännön mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
On ilmoitettu fistelimuodostustapauksia, toisinaan tuumorinekroosiin ja -regressioon liittyviä, joista osa on johtanut kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sunitinibilla hoidetuilla potilailla on raportoitu leukaluun osteonekroosia. Suurin osa tapauksista ilmeni potilailla, joilla oli leukaluun osteonekroosin tunnettuja riskitekijöitä, erityisesti altistus laskimonsisäisesti annetuille bisfosfonaateille ja/tai aikaisempia invasiivisia hoitotoimenpiteitä vaatineita hammassairauksia (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tutkimukset
Tiedot ei-kliinisistä (in vitro ja in vivo) tutkimuksista, joissa käytettiin ihmiselle suositeltua suurempia annoksia, osoittavat, että sunitinibi pystyy estämään sydämen aktiopotentiaalin repolarisaatiota (johtaen esim. QT-ajan pitenemiseen).
QTc-aika piteni yli 500 ms:iin 0,5 %:lla tutkimushenkilöistä, ja QT-aika muuttui lähtötilanteesta yli 60 ms 1,1 %:lla niistä 450 potilaasta, joilla oli kiinteä kasvain. Näitä molempia parametrejä pidetään mahdollisesti merkittävinä muutoksina. Sunitinibin on osoitettu pidentävän QTcF-aikaa (Friderician menetelmällä korjattu QT-aika) pitoisuuksilla, jotka ovat hoitopitoisuuksiin nähden noin kaksinkertaisia.
QTc-ajan pitenemistä on analysoitu 24 potilaan tutkimuksessa. Potilaat olivat 20–87-vuotiaita ja heillä oli pitkälle edennyt syöpä. Tämän tutkimuksen tulokset osoittivat, että sunitinibi vaikutti QTc-aikaan (määritelmä: kun keskimääräinen lumeryhmän tuloksella korjattu muutos oli > 10 ms, 90 %:n luottamusvälin [CI] yläraja > 15 ms) hoitopitoisuuksilla (päivä 3), kun lähtöarvoina käytettiin saman päivän ennen annostelua saatuja arvoja (within-day baseline correction). Sunitinibi vaikutti QTc- aikaan myös hoitopitoisuuden ylittävillä pitoisuuksilla (päivä 9), kun käytössä olivat molemmat lähtötilanteen korjausmenetelmät. Yhdenkään potilaan QTc-arvo ei ollut > 500 ms. Tämän löydöksen kliininen merkitys on epäselvä, vaikka vaikutus QTcF-aikaan havaittiinkin päivänä kolme 24 tuntia annostelun jälkeen (eli ajankohdassa, jolloin odotettavasti saavutetaan hoitopitoisuus plasmassa suositellulla 50 mg:n aloitusannoksella) käytettäessä päiväkohtaista korjausmenetelmää.
Kun potilaita arvioitiin kattavilla perättäisillä EKG-mittauksilla ajankohtina, jolloin sunitinibilla oli saavutettu hoitopitoisuus tai hoitopitoisuuden ylittävä pitoisuus, yhdellekään potilaalle arvioitavissa olevassa ryhmässä tai hoitoaikeen mukaisessa (ITT) -ryhmässä ei kehittynyt vaikeaksi (eli vähintään vaikeusaste 3, Common Terminology Criteria for Adverse Events [CTCAE] versio 3.0) luokiteltavaa QTc-ajan pitenemistä.
Plasman hoitopitoisuuksilla suurin keskimääräinen QTcF-ajan (Friderician korjausmenetelmä) muutos lähtötilanteesta oli 9 ms (90 % CI: 15,1 ms). Kun pitoisuudet olivat noin kaksinkertaiset, suurin QTcF- ajan muutos lähtötilanteesta oli 15,4 ms (90 % CI: 22,4 ms). Positiivisena vertailuaineena käytetyllä moksifloksasiinilla (400 mg) suurin keskimääräinen QTcF-ajan muutos lähtötilanteesta oli 5,6 ms. QTc-ajan piteneminen ei ylittänyt vaikeusastetta 2 (CTCAE versio 3.0) yhdelläkään tutkimushenkilöllä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pitkän aikavälin turvallisuus MRCC:ssä
Sunitinibin pitkän aikavälin turvallisuutta analysoitiin 9 päättyneestä tutkimuksesta, joissa sunitinibia käytettiin MRCC-potilaiden ensilinjan hoitona tai bevasitsumabi- tai sytokiinirefraktaarissa taudissa. Tutkimuksissa oli mukana yhteensä 5 739 potilasta, joista 807 (14 %) sai hoitoa vähintään 2 vuotta ja enintään 6 vuotta. Pitkäaikaista sunitinibihoitoa saaneella 807 potilaalla useimmat hoitoon liittyneet haittatapahtumat ilmenivät alun perin ensimmäisten 6 kuukauden–1 vuoden aikana, minkä jälkeen haittatapahtumien esiintyvyys vakiintui tai väheni ajan myötä. Poikkeuksena oli kilpirauhasen vajaatoiminta, jonka esiintyvyys lisääntyi vähitellen ajan kuluessa ja uusia tapauksia ilmeni koko 6 vuoden ajanjaksolla. Pitkäaikaiseen sunitinibihoitoon ei näyttänyt liittyvän uudentyyppisiä hoitoon liittyneitä haittapahtumia.
Pediatriset potilaat
Sunitinibin turvallisuusprofiili perustuu ensimmäisen vaiheen annosmääritystutkimukseen, toisen vaiheen avoimeen tutkimukseen, ensimmäisen/toisen vaiheen yksihaaraiseen tutkimukseen ja julkaisuihin, kuten seuraavassa on kuvattu.
Ensimmäisen vaiheen tutkimuksessa suun kautta otettavan sunitinibiannoksen määrittämiseksi oli mukana 35 potilasta. Näistä 30 oli lapsipotilaita (ikä: 3−17 vuotta) ja 5 nuoria aikuispotilaita (ikä: 18– 21 vuotta), joilla oli refraktaarisia kiinteitä kasvaimia. Suurimmalla osalla potilaista ensisijaisena diagnoosina oli aivokasvain. Kaikilla tutkimukseen osallistuneilla potilailla ilmeni haittavaikutuksia; useimmat haitat olivat vaikeita (vaikeusaste ≥ 3) ja näihin lukeutui sydäntoksisuus. Yleisimmät haittavaikutukset olivat ruoansulatuskanavaan kohdistuva toksisuus, neutropenia, väsymys ja ALAT-arvon nousu. Sydämeen kohdistuvien haittavaikutusten riski näytti olevan suurempi pediatrisilla potilailla, jotka ovat aikaisemmin altistuneet sydämen alueelle annetulle sädehoidolle tai antrasykliineille, kuin näille tekijöille altistumattomilla lapsipotilailla. Näillä pediatrisilla potilailla, jotka eivät aikaisemmin ole altistuneet antrasykliineille tai sydämen alueelle annetulle sädehoidolle, on tunnistettu suurin siedetty annos (maximum tolerated dose, MTD) (ks. kohta Farmakodynamiikka).
Toisen vaiheen avoimessa tutkimuksessa oli mukana 29 potilasta, joista 27 oli lapsipotilaita (ikä: 3–16 vuotta) ja 2 nuoria aikuispotilaita (ikä: 18–19 vuotta). Potilailla oli uusiutuva/etenevä/refraktaarinen korkea-asteinen gliooma tai ependymooma. Kummassakaan ryhmässä ei ilmennyt vaikeusasteen 5 haittavaikutuksia. Yleisimmät (≥ 10 %) hoitoon liittyneet haittatapahtumat olivat neutrofiilimäärän pieneneminen (6 potilaalla [20,7 %]) ja kallonsisäinen verenvuoto (3 potilaalla [10,3 %]).
Ensimmäisen/toisen vaiheen yksihaaraisessa tutkimuksessa oli mukana 6 pediatrista potilasta (ikä: 13–16 vuotta), joilla oli edennyt inoperaabeli GIST. Yleisimmät haittavaikutukset olivat ripuli, pahoinvointi, valkosolumäärän pieneneminen, neutropenia ja päänsärky. Kutakin haittaa ilmeni 3 potilaalla (50,0 %) ja ne olivat pääasiassa vaikeusastetta 1 tai 2. Neljällä potilaalla 6:sta (66,7 %) ilmeni hoitoon liittyneitä vaikeusasteen 3–4 haittatapahtumia (vaikeusasteen 3 hypofosfatemiaa, neutropeniaa ja trombosytopeniaa kutakin yhdellä potilaalla ja vaikeusasteen 4 neutropeniaa yhdellä potilaalla). Tässä tutkimuksessa ei raportoitu vakavia haittatapahtumia (SAE) eikä vaikeusasteen 5 haittavaikutuksia. Sekä tässä kliinisessä tutkimuksessa että julkaisuissa turvallisuusprofiili oli yhdenmukainen aikuisten turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Sunitinibin yliannostukselle ei ole spesifistä vastalääkettä, joten potilaalle tulee antaa yleistä elintoimintoja tukevaa hoitoa. Imeytymätön lääkeaine voidaan tarvittaessa poistaa elimistöstä oksennuttamalla tai mahahuuhtelulla. Yliannostustapauksia on raportoitu. Joihinkin tapauksiin liittyi sunitinibin tunnetun turvallisuusprofiilin mukaisia haittavaikutuksia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet, Muut proteiinikinaasin estäjät, ATC-koodi: L01EX01
Vaikutusmekanismi
Sunitinibi estää useita reseptorityrosiinikinaaseja (RTK), jotka osallistuvat kasvaimen kasvuun, neoangiogeneesiin ja syövän metastasointiin. Sunitinibi on tunnistettu lääkeaineeksi, joka estää verihiutalekasvutekijän reseptoreita (PDGFRα ja PDGFRβ), verisuonen endoteelikasvutekijän reseptoreita (VEGFR1, VEGFR2 ja VEGFR3), kantasolutekijän reseptoria (KIT), Fms-tyyppistä tyrosiinikinaasi-3:a (FLT3), kantasoluryhmiä stimuloivan kasvutekijän reseptoria (CSF-1R) ja gliasolulinjasta saadun neurotrofisen tekijän reseptoria (RET). Sunitinibin päämetaboliitin voimakkuus on ollut biokemiallisissa ja solukokeissa samaa luokkaa kuin sunitinibilla.
Kliininen teho ja turvallisuus
Sunitinibin kliinistä turvallisuutta ja tehoa on tutkittu GIST-potilailla, jotka olivat resistenttejä imatinibille (sairaus eteni imatinibihoidon aikana tai sen jälkeen); potilailla, jotka olivat intolerantteja imatinibille (imatinibihoidon aikana ilmennyt merkittävä toksisuus esti imatinibin käytön jatkamisen); MRCC-potilailla ja potilailla, joilla oli inoperaabeli haiman NET.
Teho perustuu GIST:ssa taudin etenemiseen kuluvaan aikaan (TTP) ja potilaiden eloonjäämisen lisääntymiseen, aiemmin hoitamattomassa MRCC:ssa progressiovapaaseen eloonjäämiseen (PFS), sytokiini-refraktaarisessa MRCC:ssa objektiivisiin hoitovasteisiin (ORR) ja haiman NET:ssa PFS:ään.
Gastrointestinaaliset stroomakasvaimet
Ensimmäiseen GISTiä koskeneeseen avoimeen annoshakututkimukseen osallistui potilaita, joiden imatinibihoito (mediaaniannos enintään 800 mg/vrk) oli resistenssin tai intoleranssin vuoksi epäonnistunut. Tutkimukseen otettiin mukaan 97 potilasta, jotka saivat erisuuruisia annoksia erilaisissa hoito-ohjelmissa: 55 potilasta sai 50 mg suositellun hoito-ohjelman mukaan (4 viikkoa hoitoa, 2 viikon tauko, ns. hoito-ohjelma 4/2).
Tässä tutkimuksessa mediaaniaika taudin etenemiseen oli 34,0 viikkoa (95 % CI: 22,0–46,0).
Kolmannen vaiheen satunnaistettuun, kaksoissokkoutettuun ja lumekontrolloituun sunitinibitutkimukseen otettiin GIST-potilaita, jotka olivat intolerantteja imatinibille tai joiden tauti oli edennyt imatinibihoidon (mediaaniannos enintään 800 mg/vrk) aikana tai sen jälkeen. Tutkimuksen 312 potilasta satunnaistettiin (2:1) saamaan suun kautta joko 50 mg sunitinibia tai lumelääkettä kerran vuorokaudessa hoito-ohjelmalla 4/2, kunnes tauti etenisi tai potilas lopettaisi tutkimuksen muusta syystä (potilaista 207 sai sunitinibia ja 105 lumelääkettä). Tehon ensisijainen päätetapahtuma oli aika taudin etenemiseen (= aika satunnaistamisesta ensimmäiseen objektiivisesti todennettuun taudin etenemishetkeen). Suunnitellun alustavan analyysin tekohetkellä mediaaniaika taudin etenemiseen oli sunitinibiryhmässä 28,9 viikkoa (95 % CI: 21,3–34,1) tutkijan arvioimana ja 27,3 viikkoa (95 % CI: 16,0–32,1) riippumattoman seurantaryhmän arvioimana, mikä on tilastollisesti merkitsevästi pitempi kuin lumelääkeryhmässä 5,1 viikkoa (95 % CI: 4,4–10,1) tutkijan arvioimana ja 6,4 viikkoa (95 % CI: 4,4–10,0) riippumattoman seurantaryhmän arvioimana. Ero kokonaiseloonjäämisessä oli tilastollisesti sunitinibin eduksi (riskitiheyksien suhde [HR]: 0,491 [95 % CI: 0,290–0,831]); kuolemanriski oli lumelääkeryhmässä 2 kertaa suurempi kuin sunitinibiryhmässä.
Koska alustava analyysi oli positiivinen tehon ja turvallisuuden suhteen, sokkoutus purettiin tutkimuksen riippumattoman tietojen seurantatoimikunnan suosituksesta ja lumehaaran potilaille tarjottiin sunitinibihoitoa.
Yhteensä 255 potilasta sai sunitinibia tutkimuksen avoimessa vaiheessa, mukaan lukien ne 99 potilasta, jotka saivat ensin lumelääkettä.
Ensisijaisten ja toissijaisten päätetapahtumien analyysit tutkimuksen avoimessa vaiheessa vahvistivat alustavassa analyysissä saadut tulokset, kuten taulukossa 2 on esitetty:
Taulukko 2. GIST-yhteenveto tehon päätetapahtumista (ITT-populaatio)
Kaksoissokkoutettu hoitoa | |||||
Mediaani (95 % CI) | Riskitiheyksien suhde | Lumelääke cross-over- ryhmä hoitob | |||
Päätetapahtuma | Sunitinibi | Lumelääke | (95 % CI) | p-arvo | |
Ensisijainen | |||||
TTP (viikkoa) | |||||
Välianalyysi | 27,3 (16,0; 32,1) | 6,4 (4,4; 10,0) | 0,329 (0,233; 0,466) | < 0,001 | - |
Lopullinen | 26,6 (16,0; 32,1) | 6,4 (4,4; 10,0 ) | 0,339 (0,244; 0,472) | < 0,001 | 10,4 (4,3; 22,0) |
Toissijainen | |||||
PFS (viikkoa)c | |||||
Välianalyysi | 24,1 (11,1; 28,3) | 6,0 (4,4; 9,9) | 0,333 (0,238; 0,467) | < 0,001 | - |
Lopullinen | 22,9 (10,9; 28,0) | 6,0 (4,4; 9,7) | 0,347 (0,253; 0,475) | < 0,001 | - |
ORR (%)d | |||||
Välianalyysi | 6,8 (3,7; 11,1) | 0 (-) | NA | 0,006 | - |
Lopullinen | 6,6 (3,8; 10,5) | 0 (-) | NA | 0,004 | 10,1 (5,0; 17,8) |
OS (viikkoja)e | |||||
Välianalyysi | - | - | 0,491 (0,290; 0,831) | 0,007 | - |
Lopullinen | 72,7 (61,3; 83,0) | 64,9 (45,7; 96,0) | 0,876 (0,679; 1,129) | 0,306 | - |
Lyhenteet: CI = luottamusväli, ITT = hoitoaikeen mukainen, NA = ei käytettävissä, ORR = objektiivinen hoitovaste, OS = kokonaiseloonjääminen, PFS = progressiovapaa eloonjääminen, TTP = aika taudin etenemiseen.
a Tulokset kaksoissokkoutetusta hoidosta on saatu ITT-populaatiosta ja keskitetyn radiologisen arvioinnin mittaustuloksia käyttäen.
b Tehoa koskevat tulokset 99 potilaalta, jotka siirtyivät lumelääkeryhmästä sunitinibiryhmään sokkoutuksen purkamisen jälkeen. Lähtötilanne asetettiin uudelleen cross-over-hetkellä, ja tehoanalyysit perustuivat tutkijan arviointiin.
c Alustavan analyysin PFS-luvut on päivitetty alkuperäisten tulosten uudelleenarvioinnin perusteella.
d Prosentuaalinen osuus potilaista, jotka saivat vasteen vahvistetusti (CI 95 %).
e Mediaania ei vielä saavutettu, koska tiedonkeruu oli kesken.
Kokonaiseloonjäämisen mediaani ITT-populaatiossa oli sunitinibihaarassa 72,7 viikkoa ja lumelääkehaarassa 64,9 viikkoa (HR: 0,876, 95 % CI: 0,679–1,129, p = 0,306). Tässä analyysissä lumelääkehaarassa on mukana ne potilaat, jotka myöhemmin saivat sunitinibihoitoa.
Aiemmin hoitamaton metastasoitunut munuaissolukarsinooma
Kolmannen vaiheen satunnaistetussa kansainvälisessä monikeskustutkimuksessa arvioitiin sunitinibin tehoa ja turvallisuutta verrattuna α-interferoniin potilailla, joilla oli aiemmin hoitamaton MRCC. 750 potilasta satunnaistettiin 1:1 saamaan joko sunitinibia toistuvissa 6 viikon hoitosykleissä (50 mg/vrk suun kautta 4 viikon ajan, mitä seurasi 2 viikon hoitotauko, hoito-ohjelma 4/2), tai α- interferonia (3 milj. yksikköä ihon alle ensimmäisellä viikolla, 6 milj. yksikköä toisella viikolla, 9 milj. yksikköä kolmannella viikolla ja sen jälkeen kolmena ei-perättäisenä päivänä joka viikko).
Hoidon keston mediaani oli 11,1 kuukautta (vaihteluväli: 0,4–46,1) sunitinibiryhmässä ja 4,1 kuukautta (vaihteluväli: 0,1–45,6) α-interferoniryhmässä. Sunitinibia saaneista potilaista 23,7 %:lla esiintyi vakavia hoitoon liittyviä haittavaikutuksia ja α-interferonia saaneista potilaista 6,9 %:lla. Silti haittavaikutusten takia hoidon keskeytti sunitinibipotilaista 20 % ja α- interferonipotilaista 23 %. Hoidon keskeytyksiä tapahtui 202 sunitinibipotilaalla (54 %) ja 141 α-interferonipotilaalla (39 %). Annoksen pienennyksiä oli 194 sunitinibipotilaalla (52 %) ja 98 α- interferonipotilaalla (27 %). Potilaita hoidettiin taudin etenemiseen saakka, tai kunnes he vetäytyivät tutkimuksesta. Tehon ensisijainen päätetapahtuma oli PFS. Suunniteltu alustava analyysi osoitti, että sunitinibilla saatava hyöty oli tilastollisesti merkitsevästi parempi kuin α-interferonilla. Tässä tutkimuksessa PFS:n mediaani oli sunitinibiryhmässä 47,3 viikkoa verrattuna 22,0 viikkoon α- interferoniryhmässä: HR oli 0,415 (95 % CI: 0,320–0,539, p-arvo < 0,001). Muut päätetapahtumat olivat ORR, OS ja turvallisuus. Keskitetty radiologinen arviointi lopetettiin ensisijaisen päätetapahtuman saavuttamisen jälkeen. Loppuanalyysissä ORR oli tutkijan arvioimana 46 % (95 % CI: 41–51 %) sunitinibihaarassa ja 12,0 % (95 % CI: 9–16 %) α-interferonihaarassa (p < 0,001).
Elinaika sunitinibiryhmässä oli pidempi kuin α-interferoniryhmässä. OS:n mediaani oli 114,6 viikkoa sunitinibihaarassa (95 % CI: 100,1–142,9) ja 94,9 viikkoa α-interferonihaarassa (95 % CI: 77,7–117,0): riskitiheyksien suhde oli 0,821 (95 % CI: 0,673–1,001, p = 0,0510, stratifioimaton log-rank).
Yhteenveto ITT-ryhmän PFS:tä ja OS:sta (määritetty keskitetyssä radiologisessa arvioinnissa) on esitetty taulukossa 3.
Taulukko 3. Aiemmin hoitamaton MRCC ̶ yhteenveto tehoa kuvaavista päätetapahtumista (ITT-populaatio)
Progressiovapaa eloonjääminen | Sunitinibi (N = 375) | α-interferoni (N = 375) |
Tauti ei edennyt, potilas elossa [n (%)] | 161 (42,9) | 176 (46,9) |
Taudin eteneminen tai kuolema [n (%)] | 214 (57,1) | 199 (53,1) |
PFS (viikkoa) | ||
Kvartaali (95 % CI) | ||
25 % | 22,7 (18,0; 34,0) | 10,0 (7,3; 10,3) |
50 % | 48,3 (46,4; 58,3) | 22,1 (17,1; 24,0) |
75 % | 84,3 (72,9; 95,1) | 58,1 (45,6; 82,1) |
Stratifioimaton analyysi | ||
Riskitiheyksien suhde (sunitinibi vs IFN-α) | 0,5268 | |
Riskitiheyksien suhteen 95 %:n CI (0,4316; 0,6430) | ||
p-arvoa | < 0,0001 | |
Kokonaiseloonjääminen | ||
Potilaan ei tiedetä kuolleen [n (%)] | 185 (49,3) | 175 (46,7) |
Havaittu kuolema [n (%)] | 190 (50,7) | 200 (53,3) |
OS (viikkoa) | ||
Kvartaali (95 % CI) | ||
25 % | 56,6 (48,7; 68,4) | 41,7 (32,6; 51,6) |
50 % | 114,6 (100,1; 142,9) | 94,9 (77,7; 117,0) |
75 % | NA (NA–NA) | NA (NA–NA) |
Stratifioimaton analyysi | ||
Riskitiheyksien suhde (sunitinibi vs IFN-α) | 0,8209 | |
Riskitiheyksien suhteen 95 %:n CI | (0,6730; 1,0013) | |
p-arvoa | 0,0510 | |
Lyhenteet: CI = luottamusväli, INF-α = α-interferoni, ITT = hoitoaikeen mukainen, N = potilaiden lukumäärä, NA = ei käytettävissä, OS = kokonaiseloonjääminen, PFS = progressiovapaa eloonjääminen.
a 2-suuntainen log-rank-testi.
Sytokiini-refraktaarinen metastasoitunut munuaissolukarsinooma
Toisen vaiheen sunitinibitutkimukseen otettiin potilaita, jotka olivat olleet refraktaarisia aiemmalle sytokiinihoidolle interleukiini 2:lla tai α-interferonilla. Tutkimuksen 63 potilasta saivat 50 mg:n sunitinibialoitusannoksen suun kautta kerran vuorokaudessa 4 perättäisen viikon ajan, minkä jälkeen pidettiin 2 viikon hoitotauko. Koko hoitosyklin kesto oli siis 6 viikkoa (hoito-ohjelma 4/2). Tehon ensisijainen päätetapahtuma oli objektiivisten hoitovasteiden määrä kriteereillä, joita käytetään kiinteisiin kasvaimiin (Response Evaluation Criteria in Solid Tumours, RECIST).
Tässä tutkimuksessa objektiivinen hoitovaste saavutettiin 36,5 %:lla potilaista (95 % CI: 24,7–49,6 %) ja TTP:n mediaani oli 37,7 viikkoa (95 % CI: 24,0–46,4).
Näiden tulosten vahvistamiseksi tehtiin avoin, yksihaarainen monikeskustutkimus, jossa arvioitiin sunitinibin tehoa ja turvallisuutta MRCC-potilailla, jotka olivat olleet refraktaarisia aiemmalle sytokiinihoidolle. 106 potilasta sai ainakin yhden 50 mg:n sunitinibiannoksen hoito-ohjelmalla 4/2.
Tässä tutkimuksessa tehon ensisijainen päätetapahtuma oli ORR. Toissijaiset päätetapahtumat olivat TTP, vasteen kesto (DR) ja OS.
ORR saavutettiin 35,8 %:lla potilaista (95 % CI: 26,8–47,5 %). DR:n ja OS:n mediaaneja ei ollut vielä saavutettu.
Haiman neuroendokriiniset kasvaimet
Toisen vaiheen supportiivisessa, avoimessa monikeskustutkimuksessa arvioitiin sunitinibin tehoa ja turvallisuutta monoterapiana annoksella 50 mg vuorokaudessa 4/2-hoito-ohjelmalla potilailla, joilla oli inoperaabeli haiman NET. Haiman saarekesolukasvainta sairastavien 66 potilaan kohortissa hoitovasteiden osuus (ensisijainen päätetapahtuma) oli 17 %.
Keskeisessä kolmannen vaiheen kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa lumekontrolloidussa monikeskustutkimuksessa sunitinibia tutkittiin monoterapiana potilailla, joilla oli inoperaabeli haiman NET.
Potilailla tuli olla todennettu, RECIST-kriteereihin perustuva taudin eteneminen edeltävän 12 kuukauden aikana. Heidät satunnaistettiin (1:1) saamaan joko 37,5 mg sunitinibia kerran vuorokaudessa ilman suunniteltua taukoa (N = 86) tai lumelääkettä (N = 85).
Ensisijainen tavoite oli verrata PFS:ää sunitinibia tai lumelääkettä saaneiden potilaiden välillä. Muita päätetapahtumia olivat OS, ORR, potilaiden raportoimat oireet ja elämänlaatu (PRO) ja lääkkeen turvallisuus.
Sunitinibi- ja lumelääkeryhmän potilaiden demografiset taustatiedot olivat vertailukelpoisia. Lisäksi 49 %:lla sunitinibiryhmän ja 52 %:lla lumelääkeryhmän potilaista oli toimimattomia kasvaimia, ja molemmissa tutkimushaaroissa 92 %:lla potilaista oli maksametastaaseja.
Somatostatiinianalogien käyttö oli tutkimuksessa sallittua.
Yhteensä 66 % sunitinibihaaran ja 72 % lumelääkehaaran potilaista oli saanut systeemistä hoitoa ennen tutkimusta. Lisäksi 24 % sunitinibipotilaista ja 22 % lumelääkepotilaista oli saanut somatostatiinianalogeja.
Sunitinibilla saavutettiin kliinisesti merkittävä hyöty tutkijoiden arvioimassa PFS:ssä lumelääkkeeseen verrattuna. Sunitinibihaarassa PFS:n mediaani oli 11,4 kuukautta ja lumelääkehaarassa 5,5 kuukautta [riskitiheyksien suhde 0,418 (95 % CI: 0,263–0,662), p-arvo = 0,0001]. Kuten taulukossa 4 on esitetty, tulokset taudin etenemisen suhteen olivat samanlaisia, kun RECIST-kriteereitä sovellettiin tutkijoiden mittaustuloksiin (johdettu vastearviointi). Riskitiheyksien suhde havaittiin suotuisaksi sunitinibille kaikissa lähtötilanteen ominaisuuksien mukaan arvioiduissa alaryhmissä, mukaan lukien analyysi edeltävien systeemisten hoitojen lukumäärästä. Sunitinibihaaran potilaista yhteensä 29 ja lumelääkehaaran potilaista yhteensä 24 eivät olleet saaneet edeltävää systeemistä hoitoa. Näiden potilaiden progressiovapaan eloonjäämisen riskitiheyksien suhde oli 0,365 (95 % CI: 0,156–0,857), p = 0,0156. Samalla tavoin 57 sunitinibihaaran potilaan (mukaan lukien yhden edeltävän systeemisen hoidon saaneet 28 potilasta ja kaksi tai useampia edeltäviä systeemisiä hoitoja saaneet 29 potilasta) ja 61 lumelääkehaaran potilaan (mukaan lukien yhden edeltävän systeemisen hoidon saaneet 25 potilasta ja kaksi tai useampia edeltäviä systeemisiä hoitoja saaneet 36 potilasta) PFS:n riskitiheyksien suhde oli 0,456 (95 % CI: 0,264–0,787), p = 0,0036.
PFS:n herkkyysanalyysi tehtiin tapauksissa, joissa taudin eteneminen perustui tutkijan raportoimaan kasvaimen koon mittaamiseen ja joissa kaikkien muiden syiden paitsi tutkimuksen päättymisen vuoksi sensoroituja koehenkilöitä pidettiin PFS-tapahtumina. Tämän analyysin avulla saatiin konservatiivinen ja ensisijaista analyysia tukeva sunitinibin hoitotehon arvio: riskitiheyksien suhde 0,507 (95 % CI: 0,350–0,733), p = 0,000193. Keskeinen haiman NET -tutkimus päätettiin ennenaikaisesti riippumattoman tutkimusta monitoroivan komitean suosituksesta, ja ensisijainen päätetapahtuma perustui tutkijan arviointiin. Näillä molemmilla tekijöillä on voinut olla vaikutusta hoitotehon arvioihin.
Tutkijan arvioon perustuvan PFS:n systemaattisen virheen poissulkemiseksi kuvista tehtiin sokkoutettu keskitetty riippumaton arviointi. Tehty arviointi tukee tutkijoiden arviointia, kuten taulukossa 4 on esitetty.
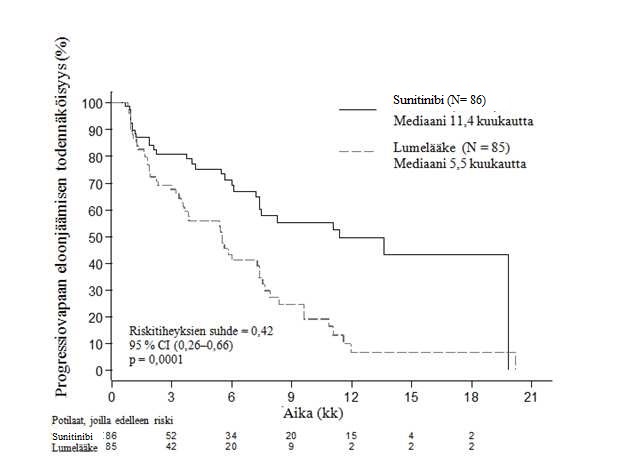
Taulukko 4. Tehoa kuvaavat tulokset kolmannen vaiheen haiman NET -tutkimuksesta
Tehoa kuvaava muuttuja | Sunitinibi (N = 86) | Lumelääke (N = 85) | Riski- tiheyksien suhde (95 % CI) | p-arvo |
Progressiovapaa eloonjääminen [mediaani, kuukautta (95 % CI)] tutkijan arvioimana | 11,4 (7,4; 19,8) | 5,5 (3,6; 7,4) | 0,418 (0,263; 0,662) | 0,0001a |
Progressiovapaa eloonjääminen [mediaani, kuukautta (95 % CI)] johdettuna vastearviointina (RECIST-kriteereitä sovellettiin tutkijoiden mittaustuloksiin) | 12,6 (7,4; 16,9) | 5,4 (3,5; 6,0) | 0,401 (0,252; 0,640) | 0,000066a |
Progressiovapaa eloonjääminen [mediaani, kuukautta (95 % CI)] sokkoutettu, riippumaton ja keskitetty kasvainten arviointi | 12,6 (11,1; 20,6) | 5,8 (3,8; 7,2) | 0,315 (0,181; 0,546) | 0,000015a |
Kokonaiseloonjääminen [5 vuoden seuranta-aika] [mediaani, kuukautta (95 % CI)] | 38,6 (25,6; 56,4) | 29,1 (16,4; 36,8) | 0,730 (0,504; 1,057) | 0,0940a |
Objektiivinen hoitovaste [%, (95 % CI)] | 9,3 (3,2; 15,4) | 0 | NA | 0,0066b |
Lyhenteet: CI = luottamusväli, N = potilaiden lukumäärä, NA = ei käytettävissä, haiman NET = haiman neuroendokriiniset kasvaimet, RECIST = vasteen arviointikriteerit kiinteissä kasvaimissa.
a 2-suuntainen stratifioimaton log-rank-testi
b Fisherin eksakti testi
Kuva 1. Kolmannen vaiheen haiman NET -tutkimuksen progressiovapaan eloonjäämisen Kaplan-Meierin kuvaaja
Lyhenteet: CI = luottamusväli, N = potilaiden lukumäärä, haiman NET = haiman neuroendokriiniset kasvaimet.
Kokonaiseloonjäämistä koskevat tulokset eivät olleet valmiit tutkimuksen päättyessä [sunitinibihaarassa 20,6 kuukautta (95 % CI: 20,6 – ei saavutettu), lumelääkehaarassa ei saavutettu (95 % CI: 15,5 – ei saavutettu), riskitiheyksien suhde 0,409 (95 % CI: 0,187–0,894), p = 0,0204]. Sunitinibihaarassa oli 9 kuolemaa ja lumelääkehaarassa 21.
Taudin edetessä potilaiden sokkoutus purettiin ja lumelääkettä saaneille potilaille tarjottiin mahdollisuutta osallistua erilliseen avoimeen sunitinibijatkotutkimukseen. Koska tutkimus päättyi ennenaikaisesti, myös tässä vaiheessa jäljellä olevien potilaiden sokkoutus purettiin ja potilaille tarjottiin mahdollisutta osallistua erilliseen avoimeen sunitinibijatkotutkimukseen. Yhteensä 59 potilasta lumelääkehaaran 85 potilaasta (69,4 %) siirtyi sunitinibihoitoon (cross-over) joko tutkimuksen aikana taudin edettyä tai tutkimuksen päättyessä, kun sokkoutus purettiin. Jatkotutkimuksessa 5 vuoden seurannan jälkeen todettu kokonaiseloonjäämisen riskitiheyksien suhde oli 0,730 (95 % CI: 0,504–1,057).
EORTC QLQ-C-30 -kyselylomakkeeseen (European Organization for Research and Treatment of Cancer Quality of Life Questionnaire) perustuvat tulokset osoittivat, että yleinen terveyteen liittyvä elämänlaatu ja 5 toiminnallista osa-aluetta (fyysinen, rooliin liittyvä, kognitiivinen, emotiaalinen ja sosiaalinen) säilyivät sunitinibipotilailla lumelääkepotilaisiin verrattuna haittavaikutusten pysyessä vähäisinä.
Neljännen vaiheen kansainvälisessä, yksihaaraisessa, avoimessa monikeskustutkimuksessa arvioitiin sunitinibin tehoa ja turvallisuutta potilailla, joilla oli etenevä, edennyt/metastasoitunut, hyvin erilaistunut, inoperaabeli haiman NET.
106 potilasta (61 potilasta aiemmin hoitamattomien kohortissa ja 45 potilasta myöhemmän hoitolinjan kohortissa) sai sunitinibia päivittäisellä annostuksella 37,5 mg kerran vuorokaudessa suun kautta ilman taukoja.
Tutkijan arvioiman progressiovapaan eloonjäämisen mediaanikesto oli 13,2 kuukautta sekä koko potilasjoukossa (95 % CI: 10,9–16,7) että aiemmin hoitamattomien kohortissa (95 % CI: 7,4–16,8).
Pediatriset potilaat
Sunitinibin käytöstä pediatrisilla potilailla on vain vähän kokemusta (ks. kohta Annostus ja antotapa).
Ensimmäisen vaiheen tutkimuksessa suun kautta otettavan sunitinibiannoksen määrittämiseksi oli mukana 35 potilasta. Näistä 30 oli lapsipotilaita (ikä: 3–17 vuotta) ja 5 nuoria aikuispotilaita (ikä: 18– 21 vuotta), joilla oli refraktaarisia kiinteitä kasvaimia. Suurimmalla osalla näistä potilaista ensisijaisena diagnoosina oli aivokasvain. Tutkimuksen ensimmäisessä osassa havaittiin annosta rajoittavaa sydäntoksisuutta. Tämän vuoksi tutkimusta muutettiin siten, että siitä suljettiin pois mahdollisesti sydäntoksista hoitoa (mukaan lukien antrasykliinejä) tai sydämen alueelle annettua sädehoitoa aikaisemmin saaneet potilaat. Tutkimuksen toisessa osassa, jossa mukana oli aiempaa syöpähoitoa saaneita potilaita, mutta joilla ei ollut sydäntoksisuuden riskitekijöitä, sunitinibihoito oli yleensä siedettyä ja kliinisesti hallittavissa annoksella 15 mg/m2 vuorokaudessa (MTD) hoito- ohjelmalla 4/2. Yksikään tutkittavista ei saanut täydellistä tai osittaista hoitovastetta. Kuudella potilaalla (17 %) todettiin stabiili tauti. Yhdellä GIST-potilaalla, joka otettiin tutkimukseen mukaan annostasolla 15 mg/m2, ei saatu näyttöä hoidon hyödystä. Havaitut haittavaikutukset olivat kaiken kaikkiaan samanlaisia kuin aikuisilla (ks. kohta Haittavaikutukset).
Toisen vaiheen avoimessa tutkimuksessa oli mukana 29 potilasta, joista 27 oli lapsipotilaita (ikä: 3– 16 vuotta) ja 2 nuoria aikuispotilaita (ikä: 18–19 vuotta). Potilailla oli korkea-asteinen gliooma tai ependymooma. Tutkimus lopetettiin tutkimussuunnitelmaan sisältyneeseen välianalyysiin, koska tauti ei pysynyt hallinnassa. PFS:n mediaani oli 2,3 kuukautta korkea-asteisen gliooman ryhmässä ja 2,7 kuukautta ependymoomaryhmässä. Kokonaiseloonjäämisen mediaani oli 5,1 kuukautta korkea-asteisen gliooman ryhmässä ja 12,3 kuukautta ependymoomaryhmässä. Yhdistettäessä em. ryhmien potilaiden tulokset yleisimmin (≥ 10 %) raportoidut hoitoon liittyneet haittatapahtumat olivat neutrofiilimäärän pieneneminen (6 potilaalla [20,7 %]) ja kallonsisäinen verenvuoto (3 potilaalla [10,3 %]) (ks. kohta Haittavaikutukset).
Näyttö ensimmäisen/toisen vaiheen tutkimuksesta, jossa oli mukana 6 pediatrista GIST-potilasta (ikä: 13–16 vuotta; sunitinibin annostus 15–30 mg/m2 vuorokaudessa suun kautta hoito-ohjelmalla 4/2), sekä saatavilla olleista julkaistuista tiedoista (20 pediatrista tai nuorta aikuista GIST-potilasta) osoitti, että sunitinibihoito johti taudin stabiloitumiseen 18 potilaalla 26:sta (69,2 %) joko imatinibihoidon epäonnistumisen tai intoleranssin kehittymisen jälkeen (16 potilaalla 21:stä stabiili tauti) tai uuden taudin kehittymisen (de novo) / leikkauksen jälkeen (2 potilaalla 5:stä stabiili tauti). Tässä ensimmäisen/toisen vaiheen tutkimuksessa stabiili tauti todettiin 3 potilaalla 6:sta (1 potilas oli saanut imatinibia neoadjuvanttihoitona) ja taudin eteneminen 3 potilaalla 6:sta (1 potilas oli saanut imatinibia adjuvanttihoitona). Samassa tutkimuksessa 4 potilaalla 6:sta (66,7 %) ilmeni hoitoon liittyneitä vaikeusasteen 3–4 haittatapahtumia (vaikeusasteen 3 hypofosfatemiaa, neutropeniaa ja trombosytopeniaa kutakin yhdellä potilaalla ja vaikeusasteen 4 neutropeniaa yhdellä potilaalla). Lisäksi julkaisuissa raportoitiin seuraavia vaikeusasteen 3 haittavaikutuksia viidellä potilaalla: väsymys (2 tapahtumaa), ruoansulatuskanavan haittavaikutukset (mukaan lukien ripuli) (2), hematologiset haittavaikutukset (mukaan lukien anemia) (2), sappirakkotulehdus (1), kilpirauhasen liikatoiminta (1) ja limakalvotulehdus (1).
Populaatiofarmakokineettisen ja farmakokineettisen/farmakodynaamisen analyysin tarkoituksena oli ekstrapoloida sunitinibin farmakokinetiikkaa sekä tärkeimpiä turvallisuuteen ja tehoon liittyviä päätetapahtumia pediatrisilla GIST-potilailla (ikä: 6–17 vuotta). Analyysi perustui tietoihin, jotka oli kerätty aikuisilta GIST-potilailta sekä aikuisilta ja lapsipotilailta, joilla oli kiinteitä kasvaimia.
Mallinnusanalyysien perusteella nuorempi ikä ja pienempi kehon koko eivät näyttäneet vaikuttavan negatiivisesti turvallisuus- ja tehovasteisiin suhteessa sunitinibialtistukseen plasmassa. Nuorempi ikä ja pienempi kehon koko eivät näyttäneet vaikuttavan negatiivisesti sunitinibin riski-hyötysuhteeseen vaan tähän vaikutti pääasiassa lääkeaineen altistus plasmassa.
EMA on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset sunitinibin käytöstä kaikkien pediatristen potilasryhmien munuais- tai munuaisallaskarsinooman hoidossa (lukuunottamatta nefroblastoomaa, nefroblastomatoosia, kirkassolusarkoomaa, mesoblastista nefroomaa, munuaisen medullaarista karsinoomaa ja munuaisen rabdoidista tuumoria) (ks. kohta Annostus ja antotapa).
EMA on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset sunitinibin käytöstä kaikkien pediatristen potilasryhmien gastroenteropankreaalisten neuroendokriinisten kasvainten hoidossa (lukuunottamatta neuroblastoomaa, neuroganglioblastomaa ja feokromosytoomaa) (ks. kohta Annostus ja antotapa).
Farmakokinetiikka
Sunitinibin farmakokinetiikkaa tutkittiin 135 terveellä vapaaehtoisella tutkimushenkilöllä ja 266 potilaalla, joilla oli kiinteä kasvain. Farmakokinetiikka oli samankaltainen kaikissa tutkituissa potilasryhmissä, joilla oli kiinteitä kasvaimia, ja terveillä vapaaehtoisilla tutkimushenkilöillä.
AUC- ja Cmax-arvot suurenevat suhteessa annokseen annosvälillä 25–100 mg. Toistuvassa päivittäisessä annostelussa sunitinibin pitoisuus 3–4-kertaistuu ja sen päämetaboliitin pitoisuus 7–10- kertaistuu. Sunitinibin ja sen aktiivisen päämetaboliitin vakaan tilan pitoisuudet saavutetaan 10–14 päivässä. Päivään 14 mennessä sunitinibin ja sen aktiivisen päämetaboliitin yhdistetty pitoisuus plasmassa on 62,9–101 ng/ml: se on prekliinisten tutkimustulosten perusteella arvioitu tavoitepitoisuusalue, joka estää reseptoreiden fosforylaation in vitro ja johtaa kasvaimen kasvun pysähtymiseen/vähenemiseen in vivo. Aktiivisen päämetaboliitin osuus kokonaispitoisuudesta on 23– 37 %. Toistuva päivittäinen anto tai toistuvat hoitosyklit testatuissa hoito-ohjelmissa eivät muuttaneet sunitinibin tai sen aktiivisen päämetaboliitin farmakokinetiikkaa merkittävästi.
Imeytyminen
Sunitinibin Cmax saavutetaan yleensä 6–12 tunnin kuluttua (aika enimmäispitoisuuden saavuttamiseen, Tmax) suun kautta annosta.
Ruokailu ei vaikuta sunitinibin biologiseen hyötyosuuteen.
Jakautuminen
In vitro -kokeissa sunitinibi sitoutui ihmisen plasman proteiineihin 95-prosenttisesti ja sen aktiivinen päämetaboliitti 90-prosenttisesti ilman ilmeistä riippuvuutta pitoisuudesta. Sunitinibin jakautumistilavuus (Vd) oli suuri (2 230 l), mikä viittaa siihen, että sunitinibi jakautuu kudoksiin.
Metaboliset interaktiot
Kaikille testatuille sytokromi P450 (CYP) -isoformeille (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 ja CYP4A9/11) lasketut in vitro Ki-arvot osoittavat, etteivät sunitinibi ja sen aktiivinen päämetaboliitti todennäköisesti indusoi kliinisesti merkittävästi sellaisten lääkeaineiden metaboliaa, jotka saattavat metaboloitua edellä mainittujen entsyymien välityksellä.
Biotransformaatio
Sunitinibi metaboloituu ensisijaisesti CYP-isoformin CYP3A4:n välityksellä aktiiviseksi päämetaboliitiksi, desetyylisunitinibiksi, jonka sama isoentsyymi metaboloi edelleen.
Sunitinibin samanaikaista antoa vahvojen CYP3A4:n induktorien tai estäjien kanssa on vältettävä, koska se voi muuttaa plasman sunitinibipitoisuuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Eliminaatio
Sunitinibi ja sen metaboliitit kulkeutuvat pääasiassa ulosteeseen (61 %). Annetusta annoksesta 16 % eliminoituu munuaisten kautta. Sunitinibi ja sen aktiivinen päämetaboliitti olivat tärkeimmät tunnistetut yhdisteet, joiden radioaktiiviset osuudet yhdistetyissä näytteissä olivat 91,5 % plasmassa, 86,4 % virtsassa ja 73,8 % ulosteessa. Vähemmän tärkeitä metaboliitteja tunnistettiin myös virtsasta ja ulosteesta mutta ei yleensä plasmasta. Kokonaispuhdistuma (CL) suun kautta annossa oli 34–62 l/h. Kun terveille vapaaehtoisille tutkimushenkilöille annettiin sunitinibia suun kautta, sen eliminaation puoliintumisaika oli noin 40–60 h ja sen tärkeimmän aktiivisen desetyylimetaboliitin 80–110 h.
Samanaikainen anto BCRP:tä estävien lääkevalmisteiden kanssa
Sunitinibi on effluksitransportteri BCRP:n substraatti in vitro. Tutkimuksessa A6181038 gefitinibin (BCRP:n estäjä) samanaikainen anto ei johtanut kliinisesti merkittävään vaikutukseen sunitinibin eikä sunitinibin + metaboliitin Cmax- ja AUC-arvoihin (ks. kohta Yhteisvaikutukset). Tämä tutkimus oli avoin vaiheen 1/2 monikeskustutkimus, jossa selvitettiin sunitinibin turvallisuutta/siedettävyyttä, suurinta siedettyä annosta ja tuumorin kasvua ehkäisevää vaikutusta MRCC-potilailla yhdistettäessä sunitinibi gefitinibiin. Tutkimuksen toissijaisena tavoittena oli tutkia gefitinibin (250 mg vuorokaudessa) ja sunitinibin (37,5 mg [kohortti 1, n = 4] tai 50 mg [kohortti 2, n = 7] vuorokaudessa 4 viikon ajan, jonka jälkeen 2 viikon tauko) yhdistelmän farmakokinetiikkaa. Sunitinibin farmakokineettisten parametrien muutokset eivät olleet kliinisesti merkittäviä eivätkä viitanneet lääkkeiden yhteisvaikutuksiin. Koska tutkimushenkilöitä oli suhteellisen vähän (n = 7 + 4) ja farmakokineettisten parametrien vaihtelu tutkimushenkilöiden välillä oli kohtalaista tai suurta, tämän tutkimuksen löydöksiä lääkkeiden farmakokineettisistä yhteisvaikutuksista on tulkittava varoen.
Erityisryhmät
Maksan toimintahäiriö
Sunitinibi ja sen päämetaboliitti metaboloituvat pääasiassa maksassa. Systeeminen altistus sunitinibin kerta-annoksen jälkeen oli samankaltainen tutkimushenkilöillä, joilla oli lievä tai keskivaikea (Child- Pughin luokat A ja B) maksan vajaatoiminta, kuin tutkimushenkilöillä, joiden maksan toiminta oli normaali. Sunitinibia ei ole tutkittu tutkimushenkilöillä, joilla maksan vajaatoiminta on vaikea (Child- Pughin luokka C).
Syöpäpotilailla tehdyistä tutkimuksista poissuljettiin ne potilaat, joiden ALAT- tai ASAT-arvo oli > 2,5 x normaali yläraja-arvo tai > 5,0 x normaali yläraja-arvo, jos syynä oli maksan etäpesäke.
Munuaisten toimintahäiriö
Populaatiofarmakokineettiset analyysit ovat osoittaneet, että sunitinibin arvioidulla kreatiniinipuhdistuman (CLcr) vaihteluvälillä (42–347 ml/min) mitattuna sunitinibin näennäinen puhdistuma (CL/F) ei muuttunut. Yksittäisen sunitinibiannoksen jälkeen systeeminen altistus oli samankaltainen vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma CLcr < 30 ml/min) sairastavilla potilailla verrattuna potilaisiin, joilla munuaisten toiminta oli normaali (CLcr > 80 ml/min). Vaikka sunitinibi ja sen päämetaboliitti eivät eliminoituneet loppuvaiheessa olevaa munuaissairautta sairastavien potilaiden hemodialyysissä, näillä potilailla systeeminen kokonaisaltistus sunitinibille oli 47 % pienempi ja sen päämetaboliitille 31 % pienempi verrattuna potilaisiin, joilla munuaisten toiminta oli normaali.
Paino, suorituskyky
Demografisten tietojen populaatiofarmakokineettiset analyysit osoittavat, ettei aloitusannosta tarvitse muuttaa potilaan ruumiinpainon tai suorituskykyä ilmaisevan ECOG (Eastern Cooperative Oncology Group) -asteen mukaan.
Sukupuoli
Saatavilla olevat tutkimustulokset viittaavat siihen, että sunitinibin näennäinen puhdistuma (CL/F) naisilla voi olla 30 % pienempi kuin miehillä. Tämä ero ei kuitenkaan vaadi annoksen muuttamista.
Pediatriset potilaat
Sunitinibin käytöstä pediatrisilla potilailla on vain vähän kokemusta (ks. kohta Annostus ja antotapa). Populaatiofarmakokineettisissä analyyseissä käytettiin yhdistettyjä tietoja aikuisista GIST-potilaista sekä aikuisista ja pediatrisista potilaista, joilla oli kiinteitä kasvaimia. Iän ja kehon koon (paino tai pinta-ala) sekä muiden selittävien muuttujien vaikutusta sunitinibin ja sen aktiivisen metaboliitin farmakokineettisiin parametreihin arvioitiin askeltavan kovariaattien mallinnusanalyysin avulla. Ikään ja kehon kokoon liittyvien testattujen muuttujien joukossa ikä oli merkittävä sunitinibin näennäiseen puhdistumaan vaikuttava muuttuja (mitä nuorempi pediatrinen potilas, sitä pienempi näennäinen puhdistuma). Samoin kehon pinta-ala oli merkittävä aktiivisen metaboliitin näennäiseen puhdistumaan vaikuttava muuttuja (mitä pienempi kehon pinta-ala, sitä pienempi näennäinen puhdistuma).
Lisäksi 3 pediatrisen tutkimuksen (2 tutkimuksessa potilailla oli kiinteitä kasvaimia ja 1 tutkimuksessa GIST; ikä: 6–11 vuotta ja 12–17 vuotta) yhdistettyjen tietojen integroidun populaatiofarmakokineettisen analyysin perusteella lähtötilanteen kehon pinta-ala (BSA) oli merkittävä sunitinibin ja sen aktiivisen metaboliitin näennäiseen puhdistumaan vaikuttava muuttuja. Tämän analyysin perusteella pediatrisille potilaille, joiden BSA on 1,10–1,87 m2, annetun noin 20 mg/m2 vuorokausiannoksen odotetaan johtavan vastaavaan (75–125 % AUC-arvosta) plasma-altistukseen sunitinibille ja sen aktiiviselle metaboliitille kuin aikuisilla GIST-potilailla, jotka saavat sunitinibia 50 mg vuorokaudessa hoito-ohjelmalla 4/2 (AUC 1233 ng.h/ml). Pediatrisissa tutkimuksissa sunitinibin aloitusannos oli 15 mg/m2 (perustuen ensimmäisen vaiheen annosmääritystutkimuksessa tunnistettuun MTD:hen, ks. kohta Farmakodynamiikka), jota suurennettiin pediatrisilla GIST-potilailla ensin annokseen 22,5 mg/m2 ja sitten annokseen 30 mg/m2 (kokonaisannos 50 mg vuorokaudessa ei saanut ylittyä) yksilöllisen turvallisuuden ja sietokyvyn perusteella. Lisäksi pediatrisia GIST-potilaita koskevissa julkaisuissa laskennallinen aloitusannos oli 16,6–36 mg/m2 ja annosta suurennettiin jopa annokseen 40,4 mg/m2 (kokonaisannos 50 mg vuorokaudessa ei ylittynyt).
Prekliiniset tiedot turvallisuudesta
Rotilla ja apinoilla tehdyissä toistuvan altistuksen toksisuustutkimuksissa, jotka kestivät pisimmillään 9 kuukautta, sunitinibilla tunnistettiin seuraavia vaikutuksia tärkeimmissä kohde-elimissä: ruoansulatuskanava (oksentelu ja ripuli apinoilla); lisämunuainen (kortikaalinen kongestio ja/tai verenvuoto rotilla ja apinoilla ja rotilla lisäksi kuolio, jota seurasi fibroosi); hemolymfopoieettinen järjestelmä (luuytimen hyposellulaarisuus ja imunestevaje kateenkorvassa, pernassa ja imusolmukkeissa); eksokriininen haima (rauhasrakkulasolujen degranulaatio, johon liittyi yksittäisten solujen kuolio); sylkirauhanen (rauhasrakkuloiden hypertrofia); nivelet (kasvulevyjen paksuneminen); kohtu (atrofia) ja munasarjat (follikkelikehityksen heikkeneminen). Kaikki löydökset ilmenivät kliinisesti merkittävillä plasman sunitinibipitoisuuksilla. Muissa tutkimuksissa todettuja muita vaikutuksia olivat QTc-ajan piteneminen, sydämen vasemman kammion ejektiofraktion pieneneminen, kivesten siementiehyiden atrofia, munuaisten mesangiaalisten solujen lisääntyminen, verenvuoto ruoansulatuskanavassa ja suun limakalvoilla ja aivolisäkkeen etuosan solujen hypertrofia. Kohdun (endometriumin atrofia) ja luiden kasvulevyjen (fyysin paksuneminen tai ruston dysplasia) muutosten arvellaan liittyvän sunitinibin farmakologiseen vaikutukseen. Useimmat näistä löydöksistä korjaantuivat 2–6 viikon kuluttua ilman hoitoa.
Genotoksisuus
Sunitinibin mahdollista genotoksisuutta on arvioitu in vitro ja in vivo. Sunitinibi ei ollut mutageeninen bakteereille, jotka oli aktivoitu metabolisesti rotan maksalla. Sunitinibi ei aiheuttanut kromosomirakenteen poikkeavuuksia ihmisen ääreisverenkierron lymfosyyttisoluissa in vitro. Sen sijaan niissä todettiin in vitro polyploidiaa (moninkertaisia kromosomipoikkeavuuksia), johon joko liittyi metabolinen aktivaatio tai ei. Sunitinibi ei ollut klastogeeninen rotan luuytimessä in vivo. Aktiivisen päämetaboliitin mahdollista genotoksisuutta ei ole arvioitu.
Karsinogeenisuus
Kuukauden kestoisessa annosmääritystutkimuksessa suun kautta letkulla annetut toistuvat vuorokausiannokset (0, 10, 25, 75 tai 200 mg/kg/vrk) rasH2-siirtogeenisille hiirille aiheuttivat karsinoomaa ja pohjukaissuolen Brunnerin rauhasten hyperplasiaa suurimmalla tutkitulla annoksella (200 mg/kg/vrk).
Kuuden kuukauden kestoinen karsinogeenisuustutkimus suoritettiin rasH2-siirtogeenisillä hiirillä, joille annettiin suun kautta letkulla toistuvia vuorokausiannoksia (0, 8, 25, 75 [pienennettiin 50:een] mg/kg/vrk). Maha-pohjukaissuolikarsinoomia, taustalla olevan hemangiosarkooman ilmaantuvuuden lisääntymistä ja/tai mahan limakalvon hyperplasiaa havaittiin annoksilla ≥ 25 mg/kg/vrk 1 tai 6 kuukauden hoidon jälkeen (≥ 7,3-kertainen AUC suositeltuja vuorokausiannoksia saaneisiin potilaisiin verrattuna).
Rotilla tehdyssä kahden vuoden kestoisessa karsinogeenisuustutkimuksessa (0, 0,33, 1 tai 3 mg/kg/vrk) sunitinibin anto 28 vuorokauden jaksoina, joita seurasi 7 vuorokauden kestoinen lääkkeetön jakso, lisäsi annoksia 3 mg/kg/vrk saavilla urosrotilla feokromosytoomien ja lisämunuaisytimen hyperplasian ilmaantumista > 1 vuoden kuluttua annostelun aloittamisesta (≥ 7,8- kertainen AUC suositeltuja vuorokausiannoksia saaneisiin potilaisiin verrattuna). Pohjukaissuolen Brunnerin rauhasten karsinoomaa ilmeni naarailla annoksella ≥ 1 mg/kg/vrk (≥ 0,9-kertainen AUC suositeltuja vuorokausiannoksia saaneisiin potilaisiin verrattuna) ja uroksilla annoksella 3 mg/kg/vrk (≥ 7,8-kertainen AUC suositeltuja vuorokausiannoksia saaneisiin potilaisiin verrattuna), ja uroksilla esiintyi limasolujen hyperplasiaa rauhasmahassa annoksella 3 mg/kg/vrk (≥ 7,8-kertainen AUC suositeltuja vuorokausiannoksia saaneisiin potilaisiin verrattuna). rasH2-siirtogeenisillä hiirillä todettujen kasvainlöydösten ja rotilla tehtyjen karsinogeenisuuslöydösten merkitystä ihmiselle ei tiedetä.
Lisääntymis- ja kehitystoksisuus
Lisääntymistoksisuustutkimuksissa ei todettu vaikutuksia urosten tai naaraiden hedelmällisyyteen. Rotille ja apinoille tehdyissä toistuvan altistuksen toksisuustutkimuksissa havaittiin, että kliinisesti merkittävä systeeminen altistus ilmeni naaraan hedelmällisyydessä follikkelin atresiana, keltarauhasen rappeutumisena, kohdun endometriumin muutoksina ja kohdun ja munasarjojen painon pienenemisenä. Kun plasman sunitinibipitoisuus oli 25-kertainen ihmisen systeemiseen altistukseen verrattuna, urosrottien hedelmällisyydessä ilmeni seuraavia vaikutuksia: kivesten siementiehyiden atrofia, lisäkivesten siittiömäärän väheneminen ja eturauhasen ja siemenrakkuloiden kolloidivaje.
Kun plasman sunitinibipitoisuus oli 5,5-kertainen ihmisen systeemiseen altistukseen verrattuna, alkio-/sikiökuolleisuus naarasrotilla ilmeni elävien sikiöiden lukumäärän merkittävänä vähenemisenä, resorptioiden lukumäärän ja implantaation jälkeisten menetysten lisääntymisenä sekä koko poikueen menetyksenä kahdeksalla tiineellä naaraalla 28:sta. Kun plasman sunitinibipitoisuus oli 3-kertainen ihmisen systeemiseen altistukseen verrattuna, tiineiden kaniinien kohdun paino pieneni ja elävien sikiöiden määrä väheni, mikä johtui resorptioiden ja implantaation jälkeisten menetysten lukumäärän lisääntymisestä sekä koko poikueen menetyksestä neljällä tiineellä naaraalla kuudesta. Rotille organogeneesin aikana annettu ≥ 5 mg/kg/vrk sunitinibihoito aiheutti kehitysvaikutuksia, kuten sikiön selkärangan epämuodostumien ilmaantuvuuden lisääntymistä. Epämuodostumat ilmenivät pääasiassa rintarangan/lannerangan luutumisen viivästymisenä ja niitä ilmeni, kun plasman sunitinibipitoisuus oli 5,5-kertainen ihmisen systeemiseen altistukseen verrattuna. Kaniineilla kehitysvaikutuksiin kuului huulihalkioiden ilmaantuvuuden lisääntyminen, kun plasman sunitinibipitoisuus oli suurin piirtein sama kuin ihmisellä havaittu. Niillä ilmeni myös huulihalkioita ja suulakihalkioita, kun plasman sunitinibipitoisuus oli 2,7-kertainen ihmisen systeemiseen altistukseen verrattuna.
Sunitinibin (0,3, 1,0, 3,0 mg/kg/vrk) vaikutuksia pre- ja postnataaliseen kehitykseen arvioitiin tiineillä rotilla tehdyssä tutkimuksessa. Emon painonnousu väheni tiineyden ja imetyksen aikana annoksilla ≥ 1 mg/kg/vrk, mutta emoon kohdistuvaa lisääntymistoksisuutta ei havaittu annoksilla 3 mg/kg/vrk saakka (arvioitu altistus ≥ 2,3-kertainen suositeltuja vuorokausiannoksia saaneiden potilaiden AUC:hen verrattuna). Jälkeläisen painon alenemista havaittiin ennen vieroitusta ja vieroituksen jälkeen annoksella 3 mg/kg/vrk. Kehitystoksisuutta ei havaittu annoksella 1 mg/kg/vrk (arvioitu altistus ≥ 0,9-kertainen suositeltuja vuorokausiannoksia saaneiden potilaiden AUC:hen verrattuna).
Farmaseuttiset tiedot
Apuaineet
12,5 mg kovat kapselit
Kapselin sisältö
Povidoni K30 LP
Mikrokiteinen selluloosa (aste 102)
Kroskarmelloosinatrium
Magnesiumstearaatti
Kapselikuori
Liivate
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Painomuste
Shellakka
Titaanidioksidi (E171)
Propyleeniglykoli
25 mg kovat kapselit
Kapselin sisältö
Povidoni K30 LP
Mikrokiteinen selluloosa (aste 102)
Kroskarmelloosinatrium
Magnesiumstearaatti
Kapselikuori
Liivate
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Painomuste
Shellakka
Titaanidioksidi (E171)
Propyleeniglykoli
50 mg kovat kapselit
Kapselin sisältö
Povidoni K30 LP
Mikrokiteinen selluloosa (aste 102)
Kroskarmelloosinatrium
Magnesiumstearaatti
Kapselikuori
Liivate
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Painomuste
Shellakka
Musta rautaoksidi (E172)
Propyleeniglykoli
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Läpipainopakkaukset: 2 vuotta.
HDPE-purkit: 2 vuotta.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SUNITINIB KRKA kapseli, kova
25 mg (L:ei) 30 x 1 fol (62,52 €)
PF-selosteen tieto
HDPE-purkki, jossa on lapsiturvallinen, kuivausainetta sisältävä polypropeeni (PP) –suljin, sisältää 30 kovaa kapselia.
OPA/Alu/PE/Alu -kerta-annosläpipainopakkaus, jossa on poistettava folio sekä kuivausainetta, sisältää 7 x 1, 10 x 1, 14 x 1, 20 x 1, 21 x 1, 28 x 1 tai 30 x 1 kovaa kapselia, kotelossa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Sunitinib Krka 12,5 mg kapseli
Kova liivatekapseli, jossa on oranssi kansiosa ja oranssi runko-osa, runko-osaan on painettu valkoisella ”SNB” ja ”12.5”. Kapseli on täytetty oranssinvärisellä jauheella. Kapselin koko: 4 (pituus noin 14 mm).
Sunitinib Krka 25 mg kapseli
Kova liivatekapseli, jossa on karamellinvärinen (vaaleanruskea) kansiosa ja oranssi runko-osa, runko-osaan on painettu valkoisella ”SNB” ja ”25”. Kapseli on täytetty oranssinvärisellä jauheella. Kapselin koko: 3 (pituus noin 16 mm).
Sunitinib Krka 50 mg kapseli
Kova liivatekapseli, jossa on karamellinvärinen (vaaleanruskea) kansiosa ja karamellinvärinen runko-osa, runko-osaan on painettu mustalla ”SNB” ja ”50”. Kapseli on täytetty oranssinvärisellä jauheella. Kapselin koko: 1EL (pidennetty; pituus noin 20 mm).
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SUNITINIB KRKA kapseli, kova
25 mg 30 x 1 fol
- Ei korvausta.
ATC-koodi
L01EX01
Valmisteyhteenvedon muuttamispäivämäärä
05.07.2021
Yhteystiedot
Tekniikantie 14
02150 Espoo
Suomi
020-7545330
www.krka.biz
info.fi@krka.biz