
TECARTUS infuusioneste, dispersio 0,4 - 2 x 10exp8 solua
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Terveydenhuollon ammattilainen
Tärkeää turvallisuustietoa terveydenhuollon ammattilaisille seuraavista riskeistä: -sytokiinioireyhtymä -vakavat neurologiset haittavaikutukset -T-soluperäiset sekundaariset syövät
Yleinen
Tärkeitä muistutuksia potilaille ja tietoa terveydenhuollon ammattilaisille.
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Tecartus (breksukabtageeniautoleuseeli) on T‑soluja sisältävä geenimuunneltu autologinen solupohjainen tuote. T‑solut on transdusoitu ex vivo käyttämällä retrovirusvektoria, joka ilmentää CD19‑molekyyliin sitoutuvaa kimeeristä antigeenireseptoria (CAR), joka muodostuu hiiren CD19‑vasta-aineen yksiketjuisesta vaihtelevasta fragmentista (scFv), joka on sitoutunut CD28‑kostimulaattoridomeeniin ja CD3‑zeeta-signalointidomeeniin.
Manttelisolulymfooma
Yhdessä potilaskohtaisessa Tecartus-valmistetta sisältävässä infuusiopussissa on breksukabtageeniautoleuseelia erän mukaan määritetyllä autologisten T‑solujen pitoisuudella. T‑solut on geenimuunneltu siten, että ne ilmentävät CD19‑molekyyliin sitoutuvaa kimeeristä antigeenireseptoria (CAR‑positiivisia elinkelpoisia T‑soluja). Lääkevalmiste on pakattu yhteen infuusiopussiin, joka sisältää kaiken kaikkiaan soludispersiota infuusiota varten tavoiteannoksella 2 x 106 CD19‑CAR‑positiivista elinkelpoista T‑solua/painokilo (vaihteluväli: 1 x 106 – 2 x 106 solua/kg), kuitenkin enintään 2 x 108 CD19‑CAR‑positiivista elinkelpoista T‑solua suspendoituna Cryostor CS10 ‑liuokseen.
Yksi infuusiopussi sisältää noin 68 ml infuusiodispersiota.
Akuutti lymfoblastinen leukemia
Yhdessä potilaskohtaisessa Tecartus-valmistetta sisältävässä infuusiopussissa on breksukabtageeniautoleuseelia erän mukaan määritetyllä autologisten T‑solujen pitoisuudella. T‑solut on geenimuunneltu siten, että ne ilmentävät CD19‑molekyyliin sitoutuvaa kimeeristä antigeenireseptoria (CAR‑positiivisia elinkelpoisia T‑soluja). Lääkevalmiste on pakattu yhteen infuusiopussiin, joka sisältää kaiken kaikkiaan soludispersiota infuusiota varten tavoiteannoksella 1 × 106 CD19‑CAR‑positiivista elinkelpoista T‑solua painokiloa kohti, kuitenkin enintään 1 x 108 CD19‑CAR‑positiivista elinkelpoista T‑solua suspendoituna Cryostor CS10 -liuokseen.
Yksi infuusiopussi sisältää noin 68 ml infuusiodispersiota.
Apuaine(et), joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 300 mg natriumia.
Yksi annos sisältää 0,05 ml dimetyylisulfoksidia (DMSO) yhtä millilitraa Tecartus-valmistetta kohden.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, dispersio.
Kliiniset tiedot
Käyttöaiheet
Manttelisolulymfooma
Tecartus on tarkoitettu aikuisille potilaille uusiutuneen tai hoitoon reagoimattoman manttelisolulymfooman (MCL) hoitoon kahden tai useamman systeemistä hoitoa, mukaan lukien Brutonin tyrosiinikinaasin (BTK) estäjää, sisältäneen hoitolinjan jälkeen.
Akuutti lymfoblastinen leukemia
Tecartus on tarkoitettu vähintään 26‑vuotiaille aikuisille potilaille uusiutuneen tai hoitoon reagoimattoman prekursori-B-solujen akuutin lymfoblastisen leukemian (ALL) hoitoon.
Ehto
CAR-T-soluhoito annetaan kvalifioidussa hoitokeskuksessa hematologisten syöpien hoitoon perehtyneen ja CAR-T-soluhoitojen antamiseen koulutetun lääkärin ohjauksessa ja valvonnassa.
Annostus ja antotapa
Tecartus täytyy antaa pätevässä kliinisessä hoitokeskuksessa. Sen saa antaa lääkäri, joka on perehtynyt hematologisten syöpien hoitoon ja saanut koulutuksen Tecartus-valmisteen antamiseen ja sillä hoidettujen potilaiden hoitamiseen. Ennen infuusiota täytyy olla ainakin yksi annos tosilitsumabia ja akuuttihoitovälineistö käytettävissä sytokiinien vapautumisoireyhtymän (cytokine release syndrome, CRS) varalta. Pätevällä hoitokeskuksella täytyy olla käytettävissä lisäannos tosilitsumabia 8 tunnin kuluessa edellisen annoksen antamisesta. Poikkeustapauksessa, jossa tosilitsumabia ei ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön takia, ennen infuusiota täytyy olla käytettävissä sopivia vaihtoehtoisia keinoja sytokiinien vapautumisoireyhtymän hoitoon tosilitsumabin sijaan.
Annostus
Tecartus on tarkoitettu vain autologiseen käyttöön (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Manttelisolulymfooma
Hoito koostuu infuusiodispersion kerta-annoksesta, jossa on CAR‑positiivisia elinkelpoisia T‑soluja yhdessä infuusiopussissa. Tavoiteannos on 2 × 106 CAR‑positiivista elinkelpoista T‑solua painokiloa kohti (vaihteluväli: 1 x 106 – 2 x 106 solua/kg) ja enimmäismäärä on vähintään 100 kg painaville potilaille 2 × 108 CAR‑positiivista elinkelpoista T‑solua.
Manttelisolulymfooman hoitoon tarkoitettu Tecartus-infuusio on suositeltavaa antaa 3–14 vuorokauden kuluttua lymfosyyttejä poistavan solunsalpaajahoidon päättymisestä. Hoidon saatavuus (eli valmisteen mahdollinen toimituspäivämäärä) täytyy varmistaa ennen lymfosyyttejä poistavaa hoitoa.
Manttelisolulymfoomapotilaiden esihoito (lymfosyyttejä poistava solunsalpaajahoito)
- Lymfosyyttejä poistava solunsalpaajahoito, jonka muodostavat syklofosfamidi 500 mg/m2laskimoon ja fludarabiini 30 mg/m2 laskimoon, on annettava ennen Tecartus-infuusiota. Suositeltavia ajankohtia ovat 5., 4. ja 3. päivä ennen Tecartus‑infuusiota.
Akuutti lymfoblastinen leukemia
Hoito koostuu infuusiodispersion kerta-annoksesta, jossa on CAR‑positiivisia elinkelpoisia T‑soluja yhdessä infuusiopussissa. Tavoiteannos on 1 × 106 CAR‑positiivista elinkelpoista T‑solua painokiloa kohti, ja enimmäisannos on vähintään 100 kg painaville potilaille 1 × 108 CAR‑positiivista elinkelpoista T‑solua.
ALL-potilaiden hoitoon tarkoitettu Tecartus-infuusio on suositeltavaa antaa 2–14 vuorokauden kuluttua lymfosyyttejä poistavan solunsalpaajahoidon päättymisestä. Hoidon saatavuus (eli valmisteen mahdollinen toimituspäivämäärä) täytyy varmistaa ennen lymfosyyttejä poistavaa hoitoa.
ALL-potilaiden esihoito (lymfosyyttejä poistava solunsalpaajahoito)
Lymfosyyttejä poistava solunsalpaajahoito, johon kuuluu 60 minuutin aikana laskimoon annettava syklofosfamidiannos 900 mg/m2, on annettava ennen Tecartus-infuusiota. Suositeltava ajankohta on 2. päivänä ennen Tecartus-infuusiota. 30 minuutin aikana laskimoon annettava fludarabiini 25 mg/m2 on annettava ennen Tecartus-infuusiota. Suositeltavia ajankohtia ovat 4., 3. ja 2. päivä ennen Tecartus‑infuusiota.
Manttelisolulymfooma ja akuutti lymfoblastinen leukemia
Esilääkitys
- Mahdollisten akuuttien infuusioreaktioiden vähentämiseksi potilaille suositellaan esilääkityksenä parasetamolia 500 – 1 000 mg suun kautta ja difenhydramiinia 12,5–25 mg laskimoon tai suun kautta (tai vastaavia lääkevalmisteita) noin 1 tunti ennen Tecartus-infuusiota.
-
Systeemisten kortikosteroidien rutiininomaista profylaktista käyttöä ei suositella (ks. kohta Yhteisvaikutukset). Systeemisten kortikosteroidien profylaktista käyttöä voidaan harkita potilaille, joilla vaikeiden immuunivälitteisten haittavaikutusten riski on suurentunut, vain siinä tapauksessa, että mahdolliset hyödyt ovat riskejä suuremmat ja käyttö on paikallisten hoitolaitoksessa noudattavien ja/tai kansallisten tai eurooppalaisten/kansainvälisten ohjeistusten mukaista.
Seuranta ennen infuusiota
- Joillakin riskialttiilla potilasryhmillä Tecartus-infuusion lykkääminen saattaa olla aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet – Syitä hoidon lykkäämiseen).
Seuranta infuusion jälkeen
- Potilasta on tarkkailtava infuusion jälkeen päivittäin ensimmäisten 7 vuorokauden ajan mahdollisen sytokiinien vapautumisoireyhtymän, neurologisten tapahtumien ja muiden toksisuuksien merkkien ja oireiden varalta. Lääkäri voi harkita sairaalahoitoa 7 vuorokauden ajaksi tai sytokiinien vapautumisoireyhtymän ja/tai neurologisten tapahtumien ensimmäisten merkkien tai oireiden ilmaantuessa.
-
Infuusion jälkeisten 7 vuorokauden jälkeen potilasta on tarkkailtava lääkärin harkinnan mukaan lisäksi vielä vähintään 7 vuorokauden ajan.
-
Potilaan on pysyteltävä pätevän hoitokeskuksen tai asianmukaisen opastuksen saaneen kliinisen hoitoyksikön läheisyydessä vähintään 2 viikon ajan infuusion jälkeen.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen vähintään 65‑vuotiailla potilailla.
Hepatiitti B -viruksen (HBV), hepatiitti C -viruksen (HCV) tai ihmisen immuunikatoviruksen (HIV) suhteen seropositiiviset potilaat
Kokemusta ei ole Tecartus-valmisteen valmistuksesta potilaille, joilla on positiivinen HIV‑testitulos tai aktiivinen HBV- tai HCV‑infektio. Näin ollen hyöty-riskisuhdetta tässä potilasryhmässä ei ole vielä varmistettu.
Pediatriset potilaat
Tecartus-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tecartus-valmisteen saa antaa vain laskimoon.
Tecartus-valmistetta ei saa säteilyttää. Valkosoluja poistavaa suodatinta EI saa käyttää.
Ennen infuusion antamista on varmistettava, että potilaan henkilöllisyys vastaa Tecartus-infuusiopussissa ja kotelossa olevia yksilöiviä potilastietoja.
Anto
- Valkosoluja poistavaa suodatinta ei saa käyttää.
- Tosilitsumabia ja ensiapuvälineitä on oltava saatavilla ennen infuusion antamista ja seurantajakson aikana. Poikkeustapauksessa, jossa tosilitsumabia ei ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön takia, ennen infuusiota täytyy olla käytettävissä sopivia vaihtoehtoisia keinoja sytokiinien vapautumisoireyhtymän hoitoon tosilitsumabin sijaan.
- Vain autologiseen käyttöön. Varmista että potilastunnus vastaa Tecartus-infuusiopussissa olevia potilaan tunnistetietoja.
- Kun infuusioletku on esitäytetty, anna Tecartus-infuusiopussin koko sisältö infuusiona 30 minuutin kuluessa joko painovoiman avulla tai peristalttisella pumpulla.
Ks. kohdasta Käyttö- ja käsittelyohjeet yksityiskohtaiset ohjeet Tecartus-valmisteen valmistelusta, annosta ja tilanteista, joissa lääkevalmisteelle on altistuttu tahattomasti, sekä Tecartus-valmisteen hävittämisestä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Lymfosyyttejä poistavan solunsalpaajahoidon vasta-aiheet täytyy ottaa huomioon.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Solupohjaisia pitkälle kehitetyssä terapiassa käytettäviä lääkkeitä (ATMP) koskevia jäljitettävyysvaatimuksia on noudatettava. Jäljitettävyyden varmistamiseksi valmisteen nimi, eränumero ja hoidettavan potilaan nimi on säilytettävä 30 vuoden ajan.
Autologinen käyttö
Tecartus on tarkoitettu ainoastaan autologiseen käyttöön, eikä sitä saa missään tilanteessa antaa muille potilaille. Ennen infuusiota on varmistettava, että potilaan henkilöllisyys vastaa Tecartus-infuusiopussissa ja kotelossa olevia potilaan tunnistetietoja. Älä anna Tecartus-infuusiota, jos potilaskohtaisen kotelon etiketin tiedot eivät vastaa kyseisen potilaan henkilöllisyyttä.
Yleistä
Lymfosyyttejä poistavan solunsalpaajahoidon varoitukset ja varotoimet täytyy ottaa huomioon.
Syitä hoidon lykkäämiseen
Tecartus-hoitoon liittyvien riskien vuoksi infuusiota on lykättävä, jos potilaalla on jokin seuraavista tiloista:
- korjautumattomat vakavat haittavaikutukset (etenkin keuhkoihin tai sydämeen liittyvät reaktiot tai matala verenpaine), mukaan lukien aiemmista solunsalpaajahoidoista johtuvat
- aktiivinen hallitsematon infektio tai tulehdussairaus
- aktiivinen käänteishyljintä (GvHD).
Joissakin tapauksissa hoitoa voidaan joutua siirtämään lymfosyyttejä poistavan solunsalpaajahoidon jälkeen. Jos infuusio siirretään yli 2 viikon päähän siitä, kun potilas on saanut lymfosyyttejä poistavaa solunsalpaajahoitoa, lymfosyyttejä poistava solunsalpaajahoito on annettava uudelleen (ks. kohta Annostus ja antotapa).
Seuranta infuusion jälkeen
Potilasta on tarkkailtava infuusion jälkeen päivittäin ensimmäisten 7 vuorokauden ajan mahdollisen sytokiinien vapautumisoireyhtymän, neurologisten tapahtumien ja muiden toksisuuksien merkkien ja oireiden varalta. Lääkäri voi harkita sairaalahoitoa 7 vuorokauden ajaksi tai sytokiinien vapautumisoireyhtymän ja/tai neurologisten tapahtumien ensimmäisten merkkien tai oireiden ilmaantuessa. Infuusion jälkeisten 7 vuorokauden jälkeen potilasta on tarkkailtava lääkärin harkinnan mukaan lisäksi vielä vähintään 7 vuorokauden ajan. Potilaille ja heitä hoitaville henkilöille on kerrottava, että sytokiinioireyhtymä tai neurologisia tapahtumia voi ilmaantua viiveellä ja että on hakeuduttava välittömästi lääkärin hoitoon, jos potilaalla ilmenee mitä tahansa sytokiinioireyhtymän tai neurologisten tapahtumien merkkejä tai oireita.
Potilaiden on pysyttävä pätevän hoitokeskuksen tai asianmukaisen opastuksen saaneen kliinisen hoitoyksikön läheisyydessä vähintään 2 viikon ajan infuusion jälkeen ja hakeuduttava välittömästi lääkärin hoitoon, jos heillä ilmenee sytokiinien vapautumisoireyhtymän merkkejä tai oireita tai neurologisia haittavaikutuksia. Jos potilaan seuranta infuusion jälkeen tapahtuu asianmukaisen opastuksen saaneessa kliinisessä hoitoyksikössä, joka ei ole hoitokeskus, hoitoyksikön on täytettävä ensihoitovälineistön ja tosilitsumabin saatavuuden suhteen samat vaatimukset kuin pätevän hoitokeskuksen. Peruselintoimintojen ja elinten toiminnan tarkkailua on harkittava reaktion vaikeusasteen mukaan.
Serologiset tutkimukset
HBV‑, HCV- ja HIV‑seulonta on tehtävä ennen solujen keräämistä Tecartus-valmisteen valmistusta varten (ks. kohta Annostus ja antotapa).
Veren, elinten, kudosten ja solujen luovutus
Tecartus-hoitoa saaneet potilaat eivät saa luovuttaa verta, elimiä, kudoksia tai soluja transplantaatiota varten.
Aktiivinen keskushermoston lymfooma
Tämän lääkevalmisteen käytöstä ei ole kokemusta potilailla, joilla on aktiivinen keskushermoston lymfooma (määritelmänä kuvantamalla varmistetut aivometastaasit). Tecartus-valmisteella hoidettiin akuuttia lymfoblastista leukemiaa sairastavia oireettomia potilaita, joilla oli enintään CNS 2 ‑luokan tauti (määritelmänä valkosolujen määrä aivo-selkäydinnesteessä < 5/µl sekä lymfoblastien esiintyminen) ilman kliinisesti todettuja neurologisia muutoksia. Tästä potilasryhmästä on kuitenkin saatavilla niukasti tietoa. Näin ollen Tecartus-valmisteen hyöty-riskisuhdetta näissä potilasryhmissä ei ole varmistettu.
Samanaikaiset sairaudet
Potilaita, joilla oli aiempi tai aktiivinen keskushermostoon liittyvä häiriö tai riittämätön munuaisten, maksan, keuhkojen tai sydämen toiminta, ei otettu mukaan tutkimuksiin. Nämä potilaat ovat todennäköisesti alttiimpia jäljempänä kuvatuille haittavaikutuksille ja tarvitsevat erityishuomiota.
Sytokiinien vapautumisoireyhtymä
Lähes kaikilla potilailla ilmeni jonkinasteinen sytokiinien vapautumisoireyhtymä. Vaikeaa sytokiinien vapautumisoireyhtymää, joka voi johtaa kuolemaan, havaittiin Tecartus-hoidon yhteydessä, ja mediaaniaika sen ilmaantumiseen oli 3 vuorokautta (vaihteluväli: 1–13 vuorokautta). Potilaita on seurattava tarkkaan näiden tapahtumien merkkien ja oireiden, kuten korkean kuumeen, hypotension, hypoksian, vilunväreiden, takykardian ja päänsäryn, varalta (ks. kohta Haittavaikutukset). Sytokiinien vapautumisoireyhtymän diagnoosi edellyttää systeemisen tulehdusreaktion vaihtoehtoisten aiheuttajien, kuten infektioiden, poissulkemista.
Tecartus-valmisteeseen liittyvän sytokiinien vapautumisoireyhtymän hoito
Ennen Tecartus-infuusiota täytyy varmistaa, että hoitokeskuksessa on käytettävissä potilasta kohti ainakin yksi annos tosilitsumabia, joka on interleukiini 6 (IL‑6) ‑reseptorin estäjä. Pätevällä hoitokeskuksella täytyy olla käytettävissä lisäannos tosilitsumabia 8 tunnin kuluessa edellisen annoksen antamisesta. Jos potilaan seuranta infuusion jälkeen tapahtuu asianmukaisen opastuksen saaneessa kliinisessä hoitoyksikössä, hoitoyksikön on täytettävä tosilitsumabin saatavuuden suhteen samat vaatimukset. Poikkeustapauksessa, jossa tosilitsumabia ei ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön takia, hoitokeskuksella / kliinisellä hoitoyksiköllä on oltava käytettävissä sopivia vaihtoehtoisia keinoja sytokiinien vapautumisoireyhtymän hoitoon tosilitsumabin sijaan.
Potilaita on hoidettava kunkin potilaan kliinisten oireiden perusteella sekä paikallisten hoitolaitoksessa noudatettavien ja/tai kansallisten tai eurooppalaisten/kansainvälisten hoitosuositusten mukaan. Lääkäreitä kehotetaan huomioimaan nämä standardit kliinisessä päätöksenteossaan.
Sytokiinien vapautumisoireyhtymän tiedetään liittyvän pääte-elinten (kuten maksan, munuaisten, sydämen ja keuhkojen) toimintahäiriöihin. Lisäksi sytokiinien vapautumisoireyhtymän yhteydessä voi esiintyä eri elimiin liittyvien perussairauksien pahenemista. Potilasta, jolla on lääketieteellisesti merkittävä sydämen toimintahäiriö, on hoidettava tehohoitostandardien ja ‑toimenpiteiden mukaisesti. Mm. sydämen kaikukuvausta on harkittava. Joissakin tapauksissa sytokiinien vapautumisoireyhtymän yhteydessä voi esiintyä makrofagiaktivaatio-oireyhtymää (MAS) ja hemofagosyyttistä lymfohistiosytoosia (HLH).
Potilaan tutkimista hemofagosyyttisen lymfohistiosytoosin tai makrofagiaktivaatio-oireyhtymän (HLH/MAS) varalta on harkittava, jos potilaalla on vaikea tai hoitoihin reagoimaton sytokiinien vapautumisoireyhtymä. Hemofagosyyttistä lymfohistiosytoosia / makrofagiaktivaatio-oireyhtymää on hoidettava paikallisen hoitolaitoksen ja/tai kansallisten tai eurooppalaisten/kansainvälisten hoitosuositusten mukaan.
Tecartus monistuu ja säilyy elimistössä tosilitsumabin ja kortikosteroidien annon jälkeen. Tuumorinekroositekijän (TNF) antagonisteja ei suositella Tecartus-valmisteeseen liittyvän sytokiinien vapautumisoireyhtymän hoidossa.
Neurologiset haittavaikutukset
Tecartus-hoitoa saaneilla potilailla on havaittu vaikeita neurologisia haittavaikutuksia, joita kutsutaan myös immuunijärjestelmän efektorisoluihin liittyväksi neurotoksisuusoireyhtymäksi (ICANS). Nämä haittavaikutukset voivat olla hengenvaarallisia tai johtaa kuolemaan. Mediaaniaika niiden ilmaantumiseen oli 7 vuorokautta (vaihteluväli: 1–262 vuorokautta) Tecartus-infuusion jälkeen (ks. kohta Haittavaikutukset).
Potilaita on hoidettava kunkin potilaan kliinisten oireiden perusteella sekä sovellettavien paikallisten hoitolaitoksessa noudatettavien ja/tai kansallisten tai eurooppalaisten/kansainvälisten hoitosuositusten mukaan. Lääkäreitä kehotetaan huomioimaan nämä standardit kliinisessä päätöksenteossaan.
Infektiot ja kuumeinen neutropenia
Vakavia infektioita, jotka voivat olla hengenvaarallisia, on havaittu hyvin yleisesti Tecartus-hoidon yhteydessä (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava infektion merkkien ja oireiden varalta ennen infuusiota, sen aikana ja sen jälkeen ja hoidettava asianmukaisesti. Profylaktisia antibiootteja on annettava hoitolaitoksen tavanomaisten ohjeiden mukaisesti.
Kuumeista neutropeniaa on havaittu potilailla Tecartus-infuusion jälkeen (ks. kohta Haittavaikutukset), ja sitä voi ilmetä samanaikaisesti sytokiinien vapautumisoireyhtymän kanssa. Kuumeisen neutropenian yhteydessä potilas on tutkittava infektion varalta ja potilasta on hoidettava laajakirjoisilla antibiooteilla, nesteillä ja muulla tukihoidolla potilaan voinnin edellyttämällä tavalla.
Immunosuppressiopotilailla on raportoitu hengenvaarallisia ja kuolemaan johtaneita opportunistisia infektioita, kuten disseminoituneita sieni-infektioita ja virusten uudelleen aktivoitumista (esim. HHV‑6 ja progressiivinen multifokaalinen leukoenkefalopatia). Tällaisten infektioiden mahdollisuus on otettava huomioon potilailla, joilla on neurologisia tapahtumia, ja on tehtävä asianmukainen diagnostinen arviointi.
Virusten reaktivoituminen
Virukset, kuten hepatiitti B ‑virus (HBV), voivat aktivoitua uudelleen potilailla, joita hoidetaan B‑soluja vastaan kohdistetuilla lääkevalmisteilla, ja aktivoituminen voi johtaa fulminanttiin hepatiittiin, maksan vajaatoimintaan ja kuolemaan.
Pitkittyneet sytopeniat
Potilailla saattaa ilmetä sytopenioita useiden viikkojen ajan lymfosyyttejä poistavan solunsalpaajahoidon ja Tecartus-infuusion jälkeen, ja heitä on hoidettava normaalien hoito-ohjeiden mukaisesti. Tecartus-infuusion jälkeen ilmeni hyvin yleisesti vähintään vaikeusasteen 3 pitkittyneitä sytopenioita, kuten trombosytopeniaa, neutropeniaa ja anemiaa (ks. kohta Haittavaikutukset). Potilaan veriarvoja on tarkkailtava Tecartus-infuusion jälkeen.
Hypogammaglobulinemia
Tecartus-hoitoa saaneilla potilailla voi ilmetä hypogammaglobulinemiaan johtavaa B‑soluaplasiaa. Tecartus-valmistetta saaneilla potilailla on havaittu hyvin yleisesti hypogammaglobulinemiaa (ks. kohta Haittavaikutukset). Hypogammaglobulinemia altistaa potilaan infektioille. Immunoglobuliinipitoisuuksia on tarkkailtava Tecartus-hoidon jälkeen, ja infektioita ehkäiseviä varotoimia, antibioottiprofylaksiaa ja uusiutuvissa infektioissa immunoglobuliinikorvaushoitoa on käytettävä normaalien hoito-ohjeiden mukaisesti.
Yliherkkyysreaktiot
Tecartus-valmisteen sisältämä DMSO tai gentamisiinijäännös saattavat aiheuttaa vakavia yliherkkyysreaktioita, kuten anafylaksiaa.
Sekundaariset syövät, mukaan luettuina T-soluperäiset sekundaariset syövät
Tecartus-hoitoa saaneille potilaille saattaa kehittyä sekundaarisia syöpiä. T-soluperäisiä syöpiä on raportoitu sen jälkeen, kun hematologisia syöpiä on hoidettu BCMA- tai CD19-kohdennetulla CAR-T-soluhoidolla. T-soluperäisiä syöpiä, myös CAR-positiivisia syöpiä, on raportoitu viikkoja ja jopa useita vuosia BCMA- tai CD19-kohdennetun CAR-T-soluhoidon jälkeen. Kuolemaan johtaneita tapauksia on esiintynyt. Potilaita on seurattava elinikäisesti sekundaaristen syöpien varalta. Jos potilaalle ilmaantuu sekundaarinen syöpä, ota yhteyttä lääkeyritykseen saadaksesi ohjeet siitä, miten potilaalta otetaan näytteet.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymää, joka saattaa olla vaikea-asteinen, on toisinaan havaittu. Tuumorilyysioireyhtymän riskin minimoimiseksi potilaiden, joiden virtsahappoarvo on koholla tai joilla on suuri kasvainkuorma, on saatava allopurinolia tai vaihtoehtoista profylaksiaa ennen Tecartus-infuusiota. Potilasta on tarkkailtava tuumorilyysioireyhtymän merkkien ja oireiden varalta, ja tapahtumia on hoidettava normaalien hoito-ohjeiden mukaisesti.
Aiempi kantasolusiirto (käänteishyljintä)
Hoidon antamista allogeenisen kantasolusiirron saaneille potilaille, joilla on aktiivinen akuutti tai krooninen käänteishyljintä, ei suositella, koska Tecartus saattaa pahentaa käänteishyljintää.
Aiempi CD19‑vasta-ainehoito
Tecartus-valmisteen käyttöä ei suositella, jos potilaan sairaus on uusiutunut CD19‑negatiivisena aiemman CD19‑vasta-ainehoidon jälkeen.
CD19‑negatiivinen akuutti lymfoblastinen leukemia
Tecartus-valmisteen käyttöä ei suositella potilaille, joilla on CD19‑negatiivinen sairaus tai joiden CD19‑statusta ei ole vahvistettu.
Natriumpitoisuus
Tämä lääkevalmiste sisältää 300 mg natriumia per infuusioannos, mikä vastaa 15 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Pitkäaikainen seuranta
On odotettavissa, että potilaat liitetään rekisteriin Tecartus-valmisteen pitkäaikaisen turvallisuuden ja tehon selvittämiseksi paremmin.
Yhteisvaikutukset
Tecartus-valmisteelle ei ole tehty yhteisvaikutustutkimuksia.
Systeemisten kortikosteroidien profylaktinen käyttö voi muuttaa Tecartus-valmisteen vaikutusta, joten niiden rutiininomaista käyttöä ennen infuusiota ei suositella (ks. kohta Annostus ja antotapa).
Eläviä taudinaiheuttajia sisältävät rokotteet
Eläviä viruksia sisältävien rokotteiden käytön turvallisuutta Tecartus-hoidon aikana tai sen jälkeen ei ole tutkittu. Varotoimena suositellaan, että eläviä viruksia sisältäviä rokotteita ei anneta ainakaan 6 viikkoon ennen lymfosyyttejä poistavaa solunsalpaajahoitoa, Tecartus-hoidon aikana eikä hoidon jälkeen ennen kuin immuniteetti on palautunut.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehillä ja naisilla
Naisille, jotka voivat tulla raskaaksi, täytyy tehdä raskaustesti mahdollisen raskauden selvittämiseksi ennen Tecartus-hoidon aloittamista.
Katso solunsalpaajahoidon tuotetiedoista tietoa tehokkaan ehkäisyn tarpeesta potilailla, jotka saavat lymfosyyttejä poistavaa solunsalpaajahoitoa.
Ei ole riittävästi altistusta koskevia tietoja, jotta voitaisiin antaa Tecartus-hoidon jälkeisen ehkäisyn kestoa koskevia suosituksia.
Raskaus
Ei ole olemassa tietoja Tecartus-valmisteen käytöstä raskaana oleville naisille. Ei ole tutkittu lisääntymis- ja kehitystoksisuutta koskevilla eläinkokeilla, voiko Tecartus-valmisteen antaminen raskaana olevalle naiselle vahingoittaa sikiötä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ei tiedetä, voiko Tecartus-valmiste siirtyä sikiöön. Jos transdusoidut solut läpäisevät istukan, ne saattavat vaikutusmekanismin perusteella aiheuttaa sikiötoksisuutta, kuten B‑lymfosytopeniaa. Siksi Tecartus-valmistetta ei suositella raskaana oleville naisille eikä naisille, jotka voivat tulla raskaaksi eivätkä käytä ehkäisyä. Raskaana oleville naisille on kerrottava mahdollisista sikiöön kohdistuvista riskeistä. Raskaudesta Tecartus-hoidon antamisen jälkeen on keskusteltava hoitavan lääkärin kanssa.
Tecartus-hoitoa saaneen äidin vastasyntyneen lapsen immunoglobuliinipitoisuuksien ja B‑solujen arviointia on harkittava.
Imetys
Ei tiedetä, erittyykö Tecartus ihmisen rintamaitoon tai siirtyykö se imetettävään lapseen. Imettävälle naiselle on kerrottava imetettävään lapseen mahdollisesti kohdistuvasta riskistä.
Hedelmällisyys
Tecartus-valmisteen vaikutuksesta hedelmällisyyteen ei ole saatavilla kliinistä tietoa. Vaikutuksia miesten ja naisten hedelmällisyyteen ei ole tutkittu eläinkokeilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tecartus-valmisteella on huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Koska neurologiset tapahtumat, mukaan lukien psyykkisen tilan muutokset tai kouristuskohtaukset, ovat mahdollisia, potilaan on vältettävä ajamista tai raskaiden tai mahdollisesti vaarallisten koneiden käyttöä vähintään 4 viikon ajan infuusion jälkeen tai kunnes neurologiset haittavaikutukset ovat hävinneet kokonaan.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Manttelisolulymfooma
Tässä kohdassa kuvatut turvallisuustiedot perustuvat Tecartus-altistukseen vaiheen 2 ZUMA‑2-tutkimuksessa, jossa 82 potilasta, joilla oli uusiutunut tai hoitoon reagoimaton manttelisolulymfooma, sai yhden annoksen CAR‑positiivisia elinkelpoisia T‑soluja (2 × 106 tai 0,5 × 106 CD19‑CAR‑T‑solua/kg) painoon perustuvan suositellun annoksen perusteella.
Merkittävimpiä ja useimmin ilmenneitä haittavaikutuksia olivat sytokiinien vapautumisoireyhtymä (CRS) (91 %), infektiot (55 %) ja enkefalopatia (51 %).
Vakavia haittavaikutuksia ilmeni 56 %:lla potilaista. Useimmin ilmenneitä vakavia haittavaikutuksia olivat enkefalopatia (26 %), infektiot (28 %) ja sytokiinien vapautumisoireyhtymä (15 %).
Vähintään vaikeusasteen 3 haittavaikutuksia ilmeni 67 %:lla potilaista. Yleisimpiä vähintään vaikeusasteen 3 muita kuin hematologisia haittavaikutuksia olivat infektiot (34 %) ja enkefalopatia (24 %). Yleisimpiä vähintään vaikeusasteen 3 hematologisia haittavaikutuksia olivat neutropenia (99 %), leukopenia (98 %), lymfopenia (96 %), trombosytopenia (65 %) ja anemia (56 %).
Akuutti lymfoblastinen leukemia
Tässä kohdassa kuvatut turvallisuustiedot perustuvat Tecartus-altistukseen vaiheen 1/2 ZUMA‑3-tutkimuksessa, jossa 100 potilasta, joilla oli uusiutunut tai hoitoon reagoimaton prekursori-B-solujen akuutti lymfoblastinen leukemia (ALL), sai yhden annoksen CAR‑positiivisia elinkelpoisia T‑soluja (0,5 × 106, 1 × 106 tai 2 x 106 CD19‑CAR‑T-solua/kg) painoon perustuvan suositellun annoksen perusteella.
Merkittävimpiä ja useimmin ilmenneitä haittavaikutuksia olivat sytokiinien vapautumisoireyhtymä (CRS) (91 %), enkefalopatia (57 %) ja infektiot (41 %).
Vakavia haittavaikutuksia ilmeni 70 %:lla potilaista. Useimmin ilmenneitä vakavia haittavaikutuksia olivat CRS (25 %), infektiot (22 %) ja enkefalopatia (21 %).
Vähintään vaikeusasteen 3 haittavaikutuksia ilmeni 76 %:lla potilaista. Yleisimpiä vähintään vaikeusasteen 3 muita kuin hematologisia haittavaikutuksia olivat infektiot (27 %), CRS (25 %) ja enkefalopatia (22 %).
Taulukkomuotoinen luettelo haittavaikutuksista
Tässä kohdassa kuvattuja haittavaikutuksia todettiin yhteensä 182:lla Tecartus-valmistetta saaneella potilaalla kahdessa keskeisessä kliinisessä monikeskustutkimuksessa: ZUMA‑2-tutkimuksessa 82:lla ja ZUMA‑3-tutkimuksessa 100 potilaalla. Haittavaikutukset on esitetty elinjärjestelmän ja esiintyvyyden mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1 Tecartus-hoidon yhteydessä todetut lääkkeen haittavaikutukset
| Elinjärjestelmä | Yleisyys | Haittavaikutukset |
|---|---|---|
| Infektiot | ||
| Hyvin yleinen | Tarkemmin määrittelemättömien patogeenien aiheuttamat infektiot Bakteeri-infektiot Sieni-infektiot Virusinfektiot | |
| Veri ja imukudos | ||
| Hyvin yleinen | Leukopeniaa Neutropeniaa Lymfopeniaa Trombosytopeniaa Anemiaa Kuumeinen neutropenia | |
| Yleinen | Koagulopatia | |
| Immuunijärjestelmä | ||
| Hyvin yleinen | Sytokiinien vapautumisoireyhtymäb Hypogammaglobulinemia | |
| Yleinen | Yliherkkyys Hemofagosyyttinen lymfohistiosytoosi | |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | Hypofosfatemiaa Heikentynyt ruokahalu Hypomagnesemia Hyperglykemiaa | |
| Yleinen | Hypoalbuminemiaa Dehydraatio | |
| Psyykkiset häiriöt | ||
| Hyvin yleinen | Delirium Ahdistuneisuus Unettomuus | |
| Hermosto | ||
| Hyvin yleinen | Enkefalopatia Vapina Päänsärky Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymä (ICANSb,c) Afasia Heitehuimaus Neuropatia | |
| Yleinen | Kouristuskohtaukset, mukaan lukien status epilepticus Ataksia Kohonnut kallonsisäinen paine | |
| Silmät | ||
| Yleinen | Näön heikentyminen | |
| Sydän | ||
| Hyvin yleinen | Takykardiat Bradykardiat | |
| Yleinen | Muu kuin kammioperäinen rytmihäiriö | |
| Verisuonisto | ||
| Hyvin yleinen | Hypotensio Hypertensio Verenvuoto | |
| Yleinen | Tromboosi | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | Yskä Dyspnea Pleuraeffuusio Hypoksia | |
| Yleinen | Hengitysvajaus Keuhkoedeema | |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | Pahoinvointi Ripuli Ummetus Vatsakipu Oksentelu Suukipu | |
| Yleinen | Suun kuivuminen Dysfagia | |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | Ihottuma Ihosairaus | |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | Tuki- ja liikuntaelimistön kipu Motorinen häiriö | |
| Munuaiset ja virtsatiet | ||
| Hyvin yleinen | Munuaisten vajaatoiminta | |
| Yleinen | Vähentynyt virtsaneritys | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | Turvotus Uupumus Kuume Kipu Vilunväristykset | |
| Yleinen | Infuusioon liittyvä reaktio | |
| Tutkimukset | ||
| Hyvin yleinen | Kohonnut alaniiniaminotransferaasiarvoa Kohonnut veren virtsahappoarvoa Kohonnut aspartaattiaminotransferaasiarvoa Hypokalsemiaa Hyponatremiaa Kohonnut konjugoituneen bilirubiinin arvoa Hypokalemiaa | |
| Yleinen | Kohonnut bilirubiiniarvoa | |
| Taulukkoon 1 otettiin mukaan vain sellaiset sytopeniat, jotka (i) aiheuttivat uusia tai pahenevia kliinisiä jälkiseurauksia, (ii) edellyttivät hoitoa tai (iii) johtivat nykyisen hoidon muuttamiseen. a Yleisyys perustuu vähintään vaikeusasteen 3 laboratorioparametreihin. b Ks. kohta Valikoitujen haittavaikutusten kuvaus. c ICANS-oireyhtymän yleisyys on arvioitu markkinoille saattamisen jälkeisen seurannan aikana ilmoitettujen tapahtumien perusteella. ZUMA‑2-tutkimuksessa viimeinen tiedonkeruupäivä: 24. heinäkuuta 2021; ZUMA‑3-tutkimuksessa viimeinen tiedonkeruupäivä: 23. heinäkuuta 2021 | ||
ZUMA‑2-tutkimuksessa ja ZUMA‑3-tutkimuksessa (n = 182) todettujen valikoitujen haittavaikutusten sekä myyntiluvan myöntämisen jälkeen raportoitujen haittavaikutusten kuvaus
Sytokiinien vapautumisoireyhtymä
Sytokiinien vapautumisoireyhtymää ilmeni 91 %:lla potilaista. Kahdellakymmenellä prosentilla (20 %) potilaista oli vähintään vaikeusasteen 3 (vaikea tai hengenvaarallinen) sytokiinien vapautumisoireyhtymä. Mediaaniaika sen ilmaantumiseen oli 3 vuorokautta (vaihteluväli: 1−13 vuorokautta), ja sen mediaanikesto oli 9 vuorokautta (vaihteluväli: 1−63 vuorokautta). Yhdeksänkymmentäseitsemän prosenttia (97 %) potilaista toipui sytokiinien vapautumisoireyhtymästä.
Yleisimpiä sytokiinien vapautumisoireyhtymään liittyviä merkkejä tai oireita potilailla, joilla oli sytokiinien vapautumisoireyhtymä, olivat kuume (94 %), hypotensio (64 %), hypoksia (32 %), vilunväristykset (31 %), takykardia (27 %), sinustakykardia (23 %), päänsärky (22 %), uupumus (16 %) ja pahoinvointi (13 %). Vakavia haittavaikutuksia, jotka saattavat liittyä sytokiinien vapautumisoireyhtymään, olivat hypotensio (22 %), kuume (15 %), hypoksia (9 %), takykardia (3 %), dyspnea (2 %) ja sinustakykardia (2 %). Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Neurologiset tapahtumat ja haittavaikutukset
Neurologisia haittavaikutuksia ilmeni 69 %:lla potilaista. Kolmellakymmenelläkahdella prosentilla (32 %) potilaista todettiin vähintään vaikeusasteen 3 (vaikeita tai hengenvaarallisia) haittavaikutuksia. Mediaaniaika haittavaikutuksen ilmaantumiseen oli 7 vuorokautta (vaihteluväli: 1−262 vuorokautta). Neurologiset tapahtumat paranivat 113 potilaalla 125:stä (90,4 %), ja niiden mediaanikesto oli 12 vuorokautta (vaihteluväli: 1−708 vuorokautta). Kolmella potilaalla oli kuollessaan neurologisia haittavaikutuksia, mukaan lukien yksi potilas, jolla raportoitiin olleen vakava enkefalopatia, ja toinen potilas, jolla raportoitiin olleen vakava sekavuustila. Loput parantumattomat neurologiset tapahtumat olivat vaikeusasteen 2 tapahtumia. Yhdeksälläkymmenelläkolmella prosentilla (93 %) kaikista hoidetuista potilaista ensimmäinen sytokiinien vapautumisoireyhtymä tai neurologinen tapahtuma ilmeni ensimmäisten 7 vuorokauden kuluessa Tecartus-infuusion antamisesta.
Yleisimmät neurologiset haittavaikutukset, ICANS mukaan lukien, olivat vapina (32 %), sekavuustila (27 %), enkefalopatia (27 %), afasia (21 %) ja kiihtyneisyys (11 %). Tecartus-valmistetta saaneilla potilailla on esiintynyt vakavia haittavaikutuksia, kuten enkefalopatiaa (15 %), afasiaa (6 %), sekavuustilaa (5 %) ja vakavaa aivoedeemaa, joka voi johtaa kuolemaan. Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Kuumeinen neutropenia ja infektiot
Kuumeista neutropeniaa on havaittu 12 %:lla potilaista Tecartus-infuusion jälkeen. Infektioita ilmeni 87 %:lla 182:sta Tectartus-valmistetta saaneesta potilaasta ZUMA‑2- ja ZUMA‑3-tutkimuksissa. Vähintään vaikeusasteen 3 (vaikeita, hengenvaarallisia tai kuolemaan johtaneita) infektioita ilmeni 30 %:lla potilaista, mukaan lukien tarkemmin määrittelemättömän patogeenin aiheuttama infektio (23 %:lla), bakteeri-infektio (8 %:lla), sieni-infektio (2 %:lla) ja virusinfektio (4 %:lla). Ks. tarkkailu- ja hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Pitkittyneet sytopeniat
Sytopeniat ovat hyvin yleisiä aiemman lymfosyyttejä poistavan solunsalpaajahoidon ja Tecartus-hoidon jälkeen.
Pitkittynyttä (päivänä 30 tai sen jälkeen jatkunutta tai päivänä 30 tai sen jälkeen alkanutta) vähintään vaikeusasteen 3 sytopeniaa ilmeni 48 %:lla potilaista, mukaan lukien neutropenia (34 %), trombosytopenia (27 %) ja anemia (15 %). Ks. hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Hypogammaglobulinemia
Hypogammaglobulinemiaa ilmeni 12 %:lla potilaista. Vähintään vaikeusasteen 3 hypogammaglobulinemiaa ilmeni 1 %:lla potilaista. Ks. hoito-ohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Immunogeenisuus
Tecartus-valmisteen immunogeenisuutta on arvioitu käyttämällä entsyymivälitteistä immunosorbenttimääritystä (ELISA) FMC63:een sitoutuvien vasta-aineiden havaitsemiseen. FMC63 on vasta-aine, josta CD19‑CAR on peräisin. Tähän mennessä CD19‑CAR-T‑solujen ei ole todettu olevan immunogeenisia manttelisolulymfoomaa sairastavilla potilailla. Hoidon alussa tehdyn seulontamäärityksen perusteella 17 ZUMA‑2-tutkimukseen osallistunutta potilasta todettiin minä tahansa testiajankohtana vasta-ainepositiivisiksi; solupohjainen ortogonaalinen varmistusmääritys kuitenkin osoitti, että kaikki ZUMA‑2-tutkimuksen 17 potilasta olivat vasta-ainenegatiivisia kaikkina testiajankohtina. Hoidon alussa tehdyn seulontamäärityksen perusteella 16 ZUMA‑3-tutkimukseen osallistunutta potilasta todettiin minä tahansa testiajankohtana vasta-ainepositiivisiksi. Niiden potilaiden joukossa, joiden näytteet olivat arvioitavissa varmistusmääritystä varten, kaksi potilasta todettiin vasta-ainepositiivisiksi hoidon jälkeen. Toisella näistä kahdesta potilaasta oli vahvistettu vasta-ainepositiivinen tulos kuukauden 6 kohdalla. Toisella potilaalla oli vahvistettu vasta-ainepositiivinen tulos uusintahoidon päivänä 28 ja kuukauden 3 kohdalla. Ei ole näyttöä siitä, että Tecartus-valmisteen alkuvaiheen ekspansion kinetiikka, CAR‑T‑solujen toiminta ja säilyminen elimistössä tai Tecartus-valmisteen turvallisuus tai teho olisi muuttunut näillä potilailla.
Sekundaariset syövät
Muilla CAR-T-soluvalmisteilla annetun hoidon jälkeen on ilmoitettu seuraavia haittavaikutuksia, joita saattaa esiintyä myös Tecartus-hoidon jälkeen: T-soluperäinen sekundaarinen syöpä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tecartus-valmisteen yliannostuksen merkeistä ei ole tietoja.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut antineoplastiset lääkeaineet, antineoplastinen solu- ja geeniterapia, ATC-koodi: L01XL06.
Vaikutusmekanismi
Tecartus, CD19‑antigeenia vastaan kohdistettuja geenimuunneltuja autologisia T‑soluja sisältävä immuunihoito, sitoutuu CD19‑antigeeniä ilmentäviin syöpäsoluihin ja normaaleihin B‑soluihin. Kun CD19‑CAR‑T‑solut tarttuvat CD19‑antigeeniä ilmentäviin kohdesoluihin, CD28‑kostimulaattoridomeeni ja CD3‑zeeta-signalointidomeeni aktivoivat alavirran puoleiset signaalikaskadit, jotka saavat T‑solut aktivoitumaan, proliferoitumaan, erilaistumaan efektorisoluiksi ja erittämään tulehdussytokiineja ja kemokiineja. Tämä tapahtumasarja johtaa CD19‑antigeeniä ilmentävien solujen kuolemaan.
Farmakodynaamiset vaikutukset
Sekä ZUMA‑2- että ZUMA‑3-tutkimuksessa arvioitiin farmakodynaamisia vasteita Tecartus-infuusion jälkeen 4 viikon aikana mittaamalla sytokiinien, kemokiinien ja muiden molekyylien pitoisuuksien tilapäistä suurenemista veressä. Sytokiinien ja kemokiinien, kuten IL‑6:n, IL‑8:n, IL‑10:n, IL‑15:n, TNF‑α:n, interferoni gamman (IFN‑γ) ja IL‑2‑reseptorin alfa-alayksikön, pitoisuudet analysoitiin. Huippupitoisuus havaittiin yleensä 8 ensimmäisen vuorokauden kuluessa infuusion jälkeen, ja pitoisuudet palautuivat yleensä lähtötasolle 28 vuorokauden kuluessa.
Tecartus-valmisteella on kohdeantigeenia ilmentäviin muihin kuin kasvainsoluihin kohdistuvia vaikutuksia, joten tilapäinen B‑soluaplasia on mahdollinen hoidon jälkeen.
Translationaalisissa analyyseissä pyrittiin tunnistamaan yhteyksiä sytokiinipitoisuuksien ja sytokiinien vapautumisoireyhtymän tai neurologisten tapahtumien ilmaantuvuuden välillä. Näissä analyyseissä osoitettiin, että useiden seerumianalyyttien, mukaan lukien IL-6:n, IL-10:n ja TNF‑α:n, suurentuneisiin pitoisuuksiin (huippupitoisuus ja AUC 1 kuukauden kohdalla) liittyi vähintään vaikeusasteen 3 neurologisia haittavaikutuksia ja vähintään vaikeusasteen 3 sytokiinien vapautumisoireyhtymää.
Kliininen teho ja turvallisuus
Uusiutunut tai hoitoon reagoimaton manttelisolulymfooma: ZUMA‑2
Vaiheen 2 yksihaaraisessa, avoimessa monikeskustutkimuksessa arvioitiin Tecartus-valmisteen tehoa ja turvallisuutta aikuisilla potilailla, joilla oli uusiutunut tai hoitoon reagoimaton manttelisolulymfooma ja jotka olivat aiemmin saaneet antrasykliiniä tai bendamustiinia sisältävää solunsalpaajahoitoa, CD20‑vasta-ainetta ja Brutonin tyrosiinikinaasin estäjää (BTKi) (ibrutinibia tai akalabrutinibia). Tutkimukseen soveltuvien potilaiden sairaus oli myös edennyt viimeisen hoidon jälkeen tai sairaus ei ollut reagoinut viimeisimpään hoitoon. Tutkimukseen eivät soveltuneet potilaat, joilla oli aktiivinen tai vakava infektio, jotka olivat aiemmin saaneet allogeenisen hematopoieettisen kantasolusiirron, joilla oli havaittavissa olevia syöpäsoluja aivo-selkäydinnesteessä tai aivometastaaseja tai joilla oli aiemmin ilmennyt keskushermoston lymfooma tai keskushermostoon liittyviä häiriöitä. ZUMA‑2-tutkimukseen otettiin mukaan yhteensä 74 potilasta (potilaat, joille tehtiin leukafereesi), ja näistä potilaista 68 sai Tecartus-hoitoa. Kolme potilasta ei saanut Tecartus-valmistetta valmistuksen epäonnistumisen takia. Kaksi muuta potilasta ei saanut hoitoa leukafereesin jälkeisen sairauden etenemisen (kuoleman) takia. Yksi potilas ei saanut Tecartus-hoitoa lymfosyyttejä poistavan solunsalpaajahoidon jälkeen jatkuneen aktiivisen eteisvärinän takia. Koko analyysipopulaatioksi (full analysis set, FAS) määritettiin kaikki potilaat, joille tehtiin leukafereesi. Yhteenveto potilaiden ominaisuuksista lähtötilanteessa on esitetty taulukossa 2.
Taulukko 2 Yhteenveto ZUMA‑2-tutkimuksen lähtötilanteen ominaisuuksista
| Luokka | Kaikki potilaat, joille tehtiin leukafereesi (FAS) |
| (N = 74) | |
| Ikä (vuotta) | |
| 65 (38, 79) |
| 58 % |
| Miessukupuoli | 84 % |
| Aiempien hoitojen määrän mediaani (minimi, maksimi) | 3 (1; 5) |
| Uusiutunutta tai hoitoon reagoimatonta sairautta sairastavien alaryhmä | |
| 42 % |
| 39 % |
| 19 % |
| Potilaat, joilla taudin levinneisyysaste oli IV | 86 % |
| Potilaat, joilla oli luuydinaffisio | 51 % |
| Morfologiset ominaisuudet | |
| 54 % |
| 26 % |
| 1 % |
| Tuntematon | 19 % |
| Saanut siltahoitoa | |
| 38 % |
| 62 % |
| Ki‑67‑immunohistokemia keskuslaboratoriossa | |
| 49 |
| 65 % |
Tecartus annettiin potilaille yhtenä infuusiona laskimoon tavoiteannoksena 2 × 106 CD19‑CAR‑T‑solua/kg (sallittu enimmäisannos: 2 × 108 solua) lymfosyyttejä poistavan solunsalpaajahoidon jälkeen. Solunsalpaajahoidon muodostivat syklofosfamidi 500 mg/m2 laskimoon ja fludarabiini 30 mg/m2 laskimoon, jotka kumpikin annettiin 5., 4. ja 3. päivänä ennen hoitoa. Leukafereesin ja lymfosyyttejä poistavan solunsalpaajahoidon välinen siltahoito solunsalpaajilla oli sallittu tautitaakan hallitsemiseksi.
Tecartus-hoitoa saaneilla potilailla mediaaniaika leukafereesista valmisteen vapauttamiseen oli 13 vuorokautta (vaihteluväli: 9−20 vuorokautta), ja mediaaniaika leukafereesista Tecartus-infuusioon oli 27 vuorokautta (vaihteluväli: 19−74 vuorokautta, lukuun ottamatta yhtä 134 vuorokauden poikkeustapausta). Mediaaniannos oli 2,0 × 106 CD19‑CAR‑T‑solua/kg. Kaikki potilaat saivat Tecartus-infuusion päivänä 0 ja olivat sairaalahoidossa vähintään päivään 7 asti.
Ensisijainen päätetapahtuma oli objektiivisen hoitovasteen saavuttaneiden potilaiden osuus (ORR) riippumattoman arviointiryhmän arvioimana Luganon 2014 kriteerien perusteella. Toissijaiset päätetapahtumat olivat vasteen kesto (DOR), kokonaiselinaika (OS), etenemisvapaa elossaoloaika (PFS) ja haittatapahtumien vaikeusaste.
Primaarianalyysin analyysijoukko määriteltiin etukäteen ja sen muodostivat 60 ensimmäistä Tecartus-hoitoa saanutta potilasta, joiden hoitovaste arvioitiin 6 kuukauden kuluttua Tecartus-infuusion jälkeisestä viikolla 4 tehdystä taudin arvioinnista. Tässä 60 potilaan analyysijoukossa objektiivisen hoitovasteen saavuttaneiden potilaiden osuus (ORR) oli 93 % ja täydellisen remission (CR) saavuttaneiden osuus oli 67 %. Objektiivisen hoitovasteen saavuttaneiden potilaiden osuus (ORR) oli merkitsevästi suurempi kuin osuus ennalta määritellyillä historiallisilla verrokeilla, 25 %, yksitahoisen merkitsevyystason ollessa 0,025 (p < 0,0001).
Tehoa koskevissa päivitetyissä 24 kuukauden seuranta-analyyseissä arvioitiin muokattua hoitoaiepopulaatiota (modified intent to treat, mITT), joka koostui 68:sta Tecartus-hoitoa saaneesta potilaasta. Kun tätä 68 potilaan mITT-populaatiota tarkasteltiin 24 kuukauden seuranta-analyysissä, objektiivisen hoitovasteen saavuttaneiden potilaiden osuus oli 91 % ja täydellisen remission saavuttaneiden osuus oli 68 %.
Taulukossa 3 esitetään koko analyysijoukkoa koskevat tulokset sekä primaarianalyysistä että 24 kuukauden seuranta-analyysistä.
Taulukko 3 Yhteenveto tehoa koskevista tuloksista ZUMA‑2-tutkimuksesta
| Luokka | Kaikki potilaat, joille tehtiin leukafereesi a (FAS) (N = 74) | |
|---|---|---|
| Primaarianalyysi | 24 kuukauden seuranta | |
| Objektiivisen hoitovasteen saavuttaneiden potilaiden osuus (ORR), n (%) [95 %:n luottamusväli] | 62 (84 %) [73,4, 91,3] | 62 (84 %) [73,4, 91,3] |
| CR n (%) [95 %:n luottamusväli] | 44 (59 %) [47,4, 70,7] | 46 (62 %) [50,1, 73,2] |
| PR n (%) [95 %:n luottamusväli] | 18 (24 %) [15,1, 35,7] | 16 (22 %) [12,9, 32,7] |
| Vasteen kesto(DOR)b | ||
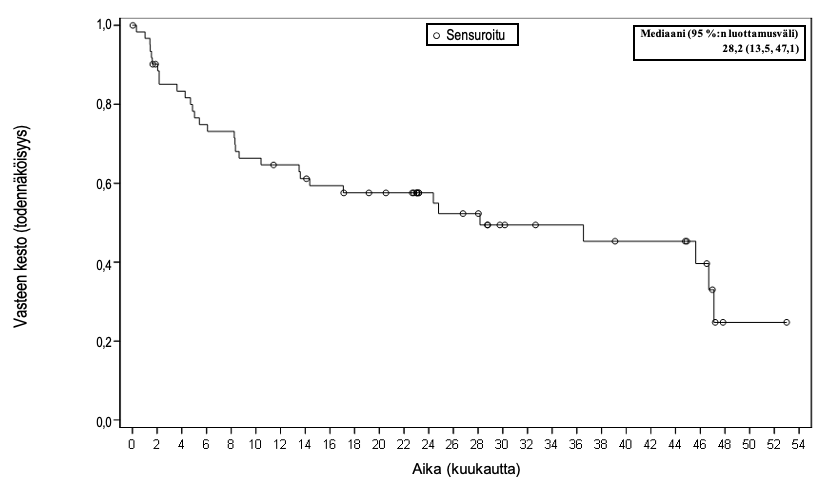
| Mediaani kuukausina [95 %:n luottamusväli] | NR [10,4, NE] | 28,2 (13,5, 47,1) |
| Vaihteluvälic kuukausina | 0,0+, 35,0+ | 0,0+, 53,0+ |
| Analyysihetkellä jatkuneet vasteet, CR+PR, CR, n (%)d | 32 (43 %), 30 (41 %) | 25 (34 %), 25 (34 %) |
| Etenemisvapaa elossaoloaika | ||
| Mediaani, kuukautta [95 %:n luottamusväli] | 16,2 [9,9, NE] | 24,0 (10,1, 48,2) |
CR, täydellinen remissio; FAS, koko analyysipopulaatio; NE, ei arvioitavissa; NR, ei saavutettu; PR, osittainen remissio. a 74:stä tutkimukseen mukaan otetusta potilaasta (potilaat, joille tehtiin leukafereesi) 69 potilasta sai lymfosyyttejä poistavaa solunsalpaajahoitoa ja 68 potilasta sai Tecartus-valmistetta. | ||
Kuva 1 Vasteen keston Kaplan–Meier‑käyrä (FAS)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Tecartus‑valmisteen käytöstä manttelisolulymfooman (MCL) hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Uusiutunut tai hoitoon reagoimaton prekursori-B-solujen akuutti lymfoblastinen leukemia (ALL): ZUMA‑3
Vaiheen 2 avoimessa monikeskustutkimuksessa arvioitiin Tecartus-valmisteen tehoa ja turvallisuutta aikuisilla potilailla, joilla oli uusiutunut tai hoitoon reagoimaton prekursori-B-solujen akuutti lymfoblastinen leukemia (ALL). Uusiutuminen tai hoitoon reagoimattomuus määriteltiin yhdeksi seuraavista: täydellistä remissiota ei ole saavutettu (primaaristi refraktaari), ensimmäinen uusiutuminen ilmeni remission jälkeen ≤ 12 kuukauden sisällä, uusiutuminen tai hoitoon reagoimattomuus ilmeni ≥ 2 hoitolinjan jälkeen, uusiutuminen tai hoitoon reagoimattomuus ilmeni allogeenisen kantasolusiirron (allo-SCT) jälkeen (edellyttäen, että siirto on tehty ≥ 100 päivää ennen tutkimukseen osallistumista ja että potilas ei ole saanut immunosuppressiivista lääkitystä ≤ 4 viikon sisällä ennen tutkimukseen osallistumista). Tutkimukseen ei otettu potilaita, joilla oli aktiivinen tai vakava infektio, aktiivinen käänteishyljintäsairaus tai aiemmin todettuja keskushermostoon liittyviä häiriöitä. Tutkimukseen hyväksyttiin potilaat, joilla oli CNS 2 -luokan tauti ilman kliinisesti todettuja neurologisia muutoksia. Vaiheen 2 ZUMA‑3-tutkimukseen otettiin mukaan yhteensä 71 potilasta (potilaat, joille tehtiin leukafereesi), ja näistä potilaista 55 sai Tecartus-hoitoa. Kuusi potilasta ei saanut Tecartus-valmistetta valmistuksen epäonnistumisen takia. Kahdeksan muuta potilasta ei saanut hoitoa ensisijaisesti leukafereesin aiheuttamien haittatapahtumien takia. Kaksi potilasta, joille tehtiin leukafereesi ja jotka saivat lymfosyyttejä poistavaa solunsalpaajahoitoa, ei saanut Tecartus-hoitoa; toisella potilaalla oli bakteremia ja kuumeinen neutropenia, ja toinen potilaista ei täyttänyt mukaanottokriteerejä lymfosyyttejä poistavan solunsalpaajahoidon jälkeen. Koko analyysipopulaatioksi (full analysis set, FAS) määritettiin kaikki potilaat, joille tehtiin leukafereesi, ja muokatuksi hoitoaiepopulaatioksi (mITT) määritettiin kaikki potilaat, joille tehtiin leukafereesi ja jotka saivat Tecartus-hoitoa vaiheessa 2. Yhteenveto potilaiden ominaisuuksista lähtötilanteessa esitetään taulukossa 4.
Taulukko 4 Yhteenveto lähtötilanteen ominaisuuksista vaiheen 2 ZUMA‑3-tutkimuksessa
| Luokka | Kaikki potilaat, joille tehtiin leukafereesi (FAS) (N = 71) | Kaikki hoidetut (mITT) (N = 55) |
|---|---|---|
| Ikä (vuotta) | ||
| 44 (19, 84) | 40 (19, 84) |
| Miessukupuoli | 58 % | 60 % |
| Valkoihoinen | 72 % | 67 % |
| Ei täydellistä remissiota (primaaristi refraktaari) | 30 % | 33 % |
| Uusiutunut/hoitoon reagoimaton tauti ≥ 2 hoitolinjan jälkeen | 76 % | 78 % |
| Ensimmäinen uusiutuminen, jos ≤ 12 kk:n sisällä ensimmäisestä remissiosta | 28 % | 29 % |
| Hoitolinjojen määrä ennen hoitoa | ||
| 2 (1, 8) | 2 (1, 8) |
| 48 % | 47 % |
| Aiemmat hoidot | ||
| 39 % | 42 % |
| 46 % | 45 % |
| 23 % | 22 % |
| Philadelphia-kromosomi (Ph+) | 27 % | 27 % |
| Allo-SCT, allogeeninen kantasolusiirto; maks., maksimi; min., minimi | ||
Lymfosyyttejä poistavan solunsalpaajahoidon jälkeen Tecartus annettiin potilaille yhtenä infuusiona laskimoon tavoiteannoksella 1 × 106 CD19‑CAR‑-T‑solua painokiloa kohti (suurin sallittu annos: 1 × 108 solua). Lymfosyyttejä poistavan solunsalpaajahoidon muodostivat 60 minuutin aikana laskimoon annettava syklofosfamidiannos 900 mg/m2 toisena päivänä ennen Tecartus-infuusiota sekä 30 minuutin aikana laskimoon annettava fludarabiiniannos 25 mg/m2, joka annettiin 4., 3. ja 2. päivänä ennen Tecartus‑infuusiota. Tecartusta saaneista 55 potilaasta 51 sai leukafereesin ja lymfosyyttejä poistavan solunsalpaajahoidon välillä siltahoitoa tautitaakan hallitsemiseksi.
Mediaaniaika leukafereesista valmisteen vapauttamiseen oli 16 vuorokautta (vaihteluväli: 11−42 vuorokautta), ja mediaaniaika leukafereesista Tecartus-infuusioon oli 29 vuorokautta (vaihteluväli: 20−60 vuorokautta). Mediaaniannos oli 1,0 × 106 CD19‑CAR‑T‑solua painokiloa kohti. Kaikki potilaat saivat Tecartus-infuusion päivänä 0 ja olivat sairaalahoidossa vähintään päivään 7 asti.
Ensijainen päätetapahtuma oli täydellisen remission kokonaisaste (overall complete remission, OCR) (täydellinen remissio [CR] + täydellinen remissio, jossa epätäydellinen hematologinen palautuminen [CRi]) riippumattoman arvioinnin perusteella potilailla, jotka saivat Tecartus-hoitoa. Kaikkien 55:n Tecartus-hoitoa saaneen potilaan joukossa (mITT-populaatio) OCR-aste oli 70,9 % ja CR-aste 56,4 % (taulukko 5), mikä oli merkittävästi suurempi kuin etukäteen määritetty kontrolliaste 40 %. Niiden 39 potilaan joukossa, jotka saavuttivat CR- tai CRi-tason, mediaaniaika vasteeseen oli 1,1 kuukautta (vaihteluväli: 0,85–2,99 kuukautta).
Kaikilla hoidetuilla potilailla oli mahdollinen seuranta-aika ≥ 18 kuukauden ajan, ja seuranta-ajan mediaani oli 20,5 kuukautta (95 %:n luottamusväli: 0,3, 32,6 kuukautta), ja kokonaiselossaolon seuranta-ajan mediaani oli 24,0 kuukautta (95 %:n luottamusväli: 23,3, 24,6).
Taulukko 5 Yhteenveto tehoa koskevista tuloksista vaiheen 2 ZUMA‑3-tutkimuksesta
| FAS N = 71 | mITTa N = 55 | |
| OCR-aste (CR + CRi) n (%) [95 % :n luottamusväli] | 39 (54,9) [43, 67] | 39 (70,9) [57,0, 82,0] |
| CR-aste, n (%) [95 %:n luottamusväli] | 31 (43,7) [32, 56] | 31 (56,4) [42,0, 70,0] |
| Jäännöstaudin (MRD) negatiivinen aste OCR (CR tai CRi) ‑potilaiden joukossa, n (%) | n = 39 38 (97 %) | n = 39 38 (97 %) |
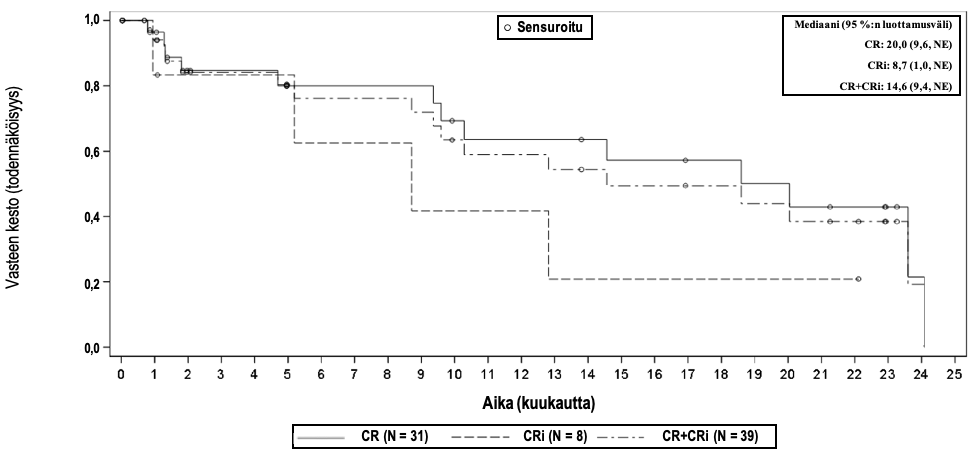
Remission kesto, mediaani kuukausina [95 %:n luottamusväli] b Vaihteluvälin mediaani kuukausina | 14,6 [9,4, NE]c (0,03+, 24,08+) | 14,6 [9,4, NE]c (0,03+, 24,08+) |
CR, täydellinen remissio; NE, ei arvioitavissa a. Tutkimukseen otetuista 71 potilaasta (joille tehtiin leukafereesi) 57 potilasta sai esihoitona solunsalpaajahoitoa ja 55 potilasta sai Tecartus-hoitoa. b. Tutkittavien viimeinen arvioitavissa oleva tautiarviointi rajattiin pois ennen uuden syöpähoidon (tyrosiinikinaasin estäjällä annetun hoidon jatkamista lukuun ottamatta) tai allo-SCT-hoidon aloittamista, jotta voitiin sulkea pois uuden hoidon mahdollinen vaikutus vasteen kestoon (DOR) tavalla, joka voisi peittää tai muuttaa KTE-X19:n vaikutusta. Tulokset analyyseistä, joissa ei käytetty rajausta seuraavan allo-SCT-hoidon tai uuden syöpähoidon mukaan, olivat yhdenmukaisia niiden analyysien kanssa, joissa tätä rajausta käytettiin. c. Remission kesto määritettiin vain OCR-tason saavuttaneilta tutkittavilta; tästä syystä FAS- ja mITT-analyysien tulokset olivat samanlaiset. | ||
Kuva 2 Kaplan–Meier-käyrä vasteen kestosta (DOR) mITT-analyysijoukossaa
a. DOR määritettiin vain OCR-tason saavuttaneilta tutkittavilta; tästä syystä FAS- ja mITT-analyysien tulokset olivat samanlaiset.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Tecartus-valmisteen käytöstä B‑solujen akuutin lymfoblastisen leukemian (ALL) hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä sekä myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Tecartus-valmisteen käytöstä alle 6 kg painavien pediatristen potilaiden hoidossa. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan.
Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa sekä manttelisolulymfooman että akuutin lymfoblastisen leukemian osalta.
Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Solukinetiikka
Manttelisolulymfooma
ZUMA‑2-tutkimuksessa todettiin annoksella 2 x 106 CD19‑CAR‑T‑solua/kg annetun Tecartus-infuusion jälkeen CD19‑CAR‑T‑soluissa nopea alkuvaiheen ekspansio, jota seurasi väheneminen lähes lähtötilanteen tasolle 3 kuukauden kuluessa. CD19‑CAR‑T‑solujen huippupitoisuudet ilmenivät ensimmäisten 7−15 vuorokauden kuluessa infuusion jälkeen.
Manttelisolulymfoomaa sairastavien potilaiden joukossa CD19‑CAR‑T‑solujen määrä veressä korreloi objektiivisen hoitovasteen (CR tai PR) kanssa (taulukko 6).
Taulukko 6 Breksukabtageeniautoleuseelisolujen farmakokinetiikka ZUMA‑2-tutkimuksessa – yhteenveto
| CD19-CAR-T-solujen määrä | Potilaat, jotka saivat hoitovasteen (CR tai PR) (N = 63) | Potilaat, jotka eivät saaneet hoitovastetta (N = 5) | P‑arvo |
| Huippu (solua/μl) Mediaani [minimi, maksimi], n | 97,52 [0,24, 2 589,47], 62 | 0,39 [0,16, 22,02], 5 | 0,0020 |
| AUC0–28 (solua/μl·päivä) Mediaani [minimi, maksimi], n | 1 386,28 [3,83–2,77 × 104], 62 | 5,51 [1,81, 293,86], 5 | 0,0013 |
P‑arvo on laskettu Wilcoxonin testillä
CD19‑CAR‑T‑solujen huippupitoisuuden mediaani manttelisolulymfooman osalta oli 74,08 solua/μl potilailla, jotka olivat ≥ 65‑vuotiaita (n = 39), ja 112,45 solua/μl potilailla, jotka olivat < 65‑vuotiaita (n = 28). CD19‑CAR‑T‑solujen AUC‑arvojen mediaani manttelisolulymfooman osalta oli 876,48 solua/μl potilailla, jotka olivat ≥ 65‑vuotiaita, ja 1 640,21 solua/μl potilailla, jotka olivat < 65‑vuotiaita.
Akuutti lymfoblastinen leukemia
ZUMA‑3-tutkimuksessa (vaihe 2) todettiin tavoiteannoksella 1 × 106 CD19‑CAR‑T-solua/kg annetun Tecartus-infuusion jälkeen CD19‑CAR‑T‑soluissa nopea alkuvaiheen ekspansio, jota seurasi väheneminen lähes lähtötilanteen tasolle 3 kuukauden kuluessa. Mediaaniaika CD19‑CAR‑T‑solujen huippupitoisuuksiin oli ensimmäisten 15 vuorokauden sisällä Tecartus-infuusion jälkeen.
Taulukossa 7 on yhteenveto Tecartus-valmisteen farmakokinetiikasta suhteessa aikaan kokonaisvasteen keskitetyn arvion perusteella.
Taulukko 7 Breksukabtageeniautoleuseelisolujen farmakokinetiikka ZUMA‑3-tutkimuksen vaiheessa 2 – yhteenveto
CD19-CAR-T-solujen määrä | Potilaat, jotka saavuttivat täydellisen remission | Potilaat, jotka eivät saavuttaneet täydellistä remissiotaa | P‑arvo |
|---|---|---|---|
Huippu (solua/μl) | 38,35 [1,31, 1 533,4], | 0,49 [0,00, 183,50], | 0,0001c |
AUC0–28 (solua/μl·päivä) | 424,03 [14,12, 19 390,42], | 4,12 [0,00, 642,25], | 0, 0001c |
a. Kolmesta tutkittavasta 39:stä, jotka saavuttivat CR- tai CRi-tason, ja kahdesta tutkittavasta 16:sta, jotka eivät saavuttaneet CR- tai CRi-tasoa, ei ollut lainkaan CD19-CAR-T-soluja koskevaa tietoa millään infuusion jälkeisellä käynnillä.
b.”Ei täydellistä remissiota” sisältää kaikki tutkittavat, jotka eivät saavuttaneet CR- tai CRi-tasoa ja joiden vasteeksi luokitellaan epätäydellinen remissio sekä osittainen hematologinen palautuminen, blastiton hypoplastinen tai aplastinen luuydin (N = 4), osittainen vaste (N = 0), ei vastetta (N = 9) tai ei arvioitavissa (N = 3).
c. p‑arvo on laskettu Wilcoxonin testillä.
CD19‑CAR‑T-solujen huippupitoisuuden mediaani akuutin lymfoblastisen leukemian (ALL) osalta oli 34,8 solua/μl potilailla, jotka olivat ≥ 65‑vuotiaita (n = 8), ja 17,4 solua/μl potilailla, jotka olivat < 65‑vuotiaita (n = 47). CD19‑CAR‑T-solujen AUC-arvojen mediaani akuutin lymfoblastisen leukemian osalta oli 425,0 solua/μl päivässä potilailla, jotka olivat ≥ 65‑vuotiaita, ja 137,7 solua/μl päivässä potilailla, jotka olivat < 65‑vuotiaita.
Manttelisolulymfoomaa ja akuuttia lymfoblastista leukemiaa sairastavien sukupuolella ei ollut merkittävää vaikutusta Tecartus-valmisteen AUCpäivä 0–28‑arvoon eikä Cmax‑arvoon.
Tutkimuksia Tecartus-valmisteen käytöstä potilaille, joilla on maksan tai munuaisten vajaatoiminta, ei ole tehty.
Prekliiniset tiedot turvallisuudesta
Tecartus-valmiste koostuu geneettisesti muunnelluista ihmisen T‑soluista, eikä siksi ole olemassa edustavia in vitro ‑analyysejä tai ex vivo- tai in vivo ‑malleja, jotka voisivat täsmällisesti kuvata tämän ihmisperäisen valmisteen toksikologisia piirteitä. Tästä syystä konventionaalisia lääkekehityksessä käytettäviä toksisuutta koskevia tutkimuksia ei ole tehty.
Karsinogeenisuutta tai genotoksisuutta koskevia tutkimuksia ei ole tehty.
Ei ole tehty tutkimuksia, joilla arvioitaisiin tämän hoidon vaikutuksia hedelmällisyyteen, lisääntymiseen ja kehitykseen.
Farmaseuttiset tiedot
Apuaineet
Cryostor CS10 (sisältää DMSO:ta)
Natriumkloridi
Ihmisen albumiini
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
1 vuosi.
Tecartus säilyy huoneenlämmössä (20−25 °C) enintään 3 tuntia sulatuksen jälkeen. Tecartus‑infuusio on kuitenkin aloitettava 30 minuutin kuluessa siitä, kun valmiste on täysin sulanut, eikä infuusion kokonaiskesto saa ylittää 30:tä minuuttia.
Säilytys
Tecartus-valmiste täytyy säilyttää ≤ ‑120 °C:ssa, ja se täytyy säilyttää pakastettuna, kunnes potilas on valmis hoitoa varten. Näin varmistetaan, että potilas saa elinkelpoisia eläviä autologisia soluja.
Tecartus-valmistetta voidaan yhden kerran säilyttää ‑80 °C:ssa (± 10 °C) enintään 90 päivän ajan. Jos valmistetta on säilytetty ‑80 °C:ssa (± 10 °C), se on käytettävä joko kyseisen 90 päivän jakson päättymiseen tai pakkaukseen merkittyyn viimeiseen käyttöpäivämäärään mennessä sen mukaan, kumpi näistä toteutuu ensin. Näiden ajankohtien jälkeen valmistetta ei saa enää käyttää, vaan se on hävitettävä.
Sulanutta lääkevalmistetta ei saa pakastaa uudelleen.
Sulatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TECARTUS infuusioneste, dispersio
0,4 - 2 x 10exp8 solua (L:ei) 1 kpl (68 ml) (408780,60 €)
PF-selosteen tieto
Etyleeni‑vinyyliasetaattikryosäilytyspussi, jossa on sulkimella varustettu lisäysletku ja kaksi käytettävissä olevaa läpäistävää porttia. Pussi sisältää noin 68 ml soludispersiota.
Yksi kryosäilytyspussi on yksittäispakattu metalliseen kuljetuskoteloon.
Valmisteen kuvaus:
Kirkas tai läpinäkymätön, valkoinen tai punainen dispersio.
Käyttö- ja käsittelyohjeet
Säteilytys saattaa inaktivoida valmisteen.
Ennen lääkkeen käsittelyä tai antoa huomioon otettavat varotoimet
Tecartus-valmiste on kuljetettava hoitoyksikön sisällä suljetuissa, hajoamattomissa, vuotamattomissa säiliöissä.
Tämä lääkevalmiste sisältää ihmisen verisoluja. Tecartus-valmistetta käsittelevien terveydenhuollon ammattilaisten on noudatettava asianmukaisia varotoimia (käytettävä suojakäsineitä ja ‑laseja) mahdollisten tartuntatautien välttämiseksi.
Valmistelut ennen antoa
- Varmista, että potilaan henkilöllisyys (henkilötunnus) vastaa Tecartus-metallikotelon potilastunnisteita.
- Tecartus-infuusiopussia ei saa poistaa metallikotelosta, jos potilaskohtaisen etiketin tiedot eivät vastaa sen potilaan tietoja, jolle hoito on tarkoitus antaa.
- Kun potilaan henkilötunnus on vahvistettu, poista infuusiopussi metallikotelosta.
- Tarkista, että metallikotelon etiketin potilastiedot vastaavat infuusiopussin etiketin tietoja.
- Tarkasta ennen valmisteen sulattamista, ettei infuusiopussi ole rikkoutunut. Jos infuusiopussi ei ole ehjä, noudata paikallista ohjeistusta ihmisperäisen jätemateriaalin käsittelystä (ja ota välittömästi yhteyttä Kiteen).
Sulatus
- Pakkaa infuusiopussi toisen pussin sisään.
- Sulata Tecartus-valmistetta noin 37 °C:ssa joko vesihauteessa tai kuivasulatusmenetelmällä, kunnes infuusiopussissa ei näy enää jäätä. Sekoita infuusiopussin sisältöä varovasti paakkuuntuneen soluaineksen hajottamiseksi. Jos näkyviä solupaakkuja on jäljellä, jatka infuusiopussin sisällön varovaista sekoittamista. Pienten soluainespaakkujen pitäisi hajota, kun valmistetta sekoitetaan varovasti käsin. Tecartus-valmistetta ei saa pestä, sentrifugoida eikä suspendoida uudelleen uuteen väliaineeseen ennen infuusiota. Sulattaminen kestää noin 3−5 minuuttia.
- Sulattamisen jälkeen Tecartus säilyy huoneenlämmössä (20−25 °C) enintään 3 tuntia. Tecartus-infuusio on kuitenkin aloitettava 30 minuutin kuluessa siitä, kun valmiste on täysin sulanut.
Anto
- Vain autologiseen kertakäyttöön.
- Tosilitsumabia ja ensiapuvälineitä on oltava saatavilla ennen infuusion antamista ja seurantajakson aikana. Poikkeustapauksessa, jossa tosilitsumabia ei ole saatavilla Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön takia, ennen infuusiota täytyy olla käytettävissä sopivia vaihtoehtoisia keinoja sytokiinien vapautumisoireyhtymän hoitoon tosilitsumabin sijaan.
- Valkosoluja poistavaa suodatinta ei saa käyttää.
- Tecartus-valmisteen antamiseen suositellaan keskuslaskimoyhteyttä.
- Varmista, että potilastunnus vastaa Tecartus-infuusiopussissa olevia potilastunnistetietoja.
- Esitäytä infuusioletku 9 mg/ml (0,9 %) natriumkloridi-injektioliuoksella (0,154 mmol natriumia millilitrassa) ennen infuusiota.
- Anna Tecartus-infuusiopussin koko sisältö infuusiona 30 minuutin kuluessa joko painovoiman avulla tai peristalttisella pumpulla.
- Sekoittele infuusiopussia varovasti infuusion aikana solujen paakkuuntumisen estämiseksi.
- Kun infuusiopussin koko sisältö on infusoitu, huuhtele laskimolinja samalla infuusionopeudella 9 mg/ml (0,9 %) natriumkloridi-injektioliuoksella (0,154 mmol natriumia millilitrassa), jotta varmistetaan, että potilas on saanut koko hoidon.
Lääkevalmisteen hävittämisessä huomioon otettavat varotoimet
Käyttämätön lääkevalmiste ja kaikki Tecartus‑valmisteen kanssa kosketuksessa olleet materiaalit (kiinteä ja nestemäinen jäte) on käsiteltävä ja hävitettävä ihmisperäisen jätemateriaalin käsittelyä koskevien paikallisten ohjeiden mukaisesti.
Tahaton altistus
Tahattoman altistuksen sattuessa on noudatettava ihmisperäisen materiaalin käsittelyä koskevia paikallisia ohjeita. Tecartus-valmisteen kanssa mahdollisesti kosketuksissa olleet työskentelytasot ja materiaalit on puhdistettava asianmukaisella desinfiointiaineella.
Korvattavuus
TECARTUS infuusioneste, dispersio
0,4 - 2 x 10exp8 solua 1 kpl
- Ei korvausta.
ATC-koodi
L01XL06
Valmisteyhteenvedon muuttamispäivämäärä
04.05.2026
Yhteystiedot
Karhumäentie 3
01530 Vantaa
Suomi
09 42726918
www.gilead.se/utility/contact
nordics.medinfo@gilead.com