CLUVOT pulver och vätska till injektions-/infusionsvätska, lösning 250 IU, 1250 IU
Tilläggsinformation
Cluvot 250 IU
Cluvot 1250 IU
Pulver och vätska till injektions-/infusionsvätska, lösning
Human koagulationsfaktor XIII
Allmänna direktiv
Läs noga igenom denna bipacksedel innan du börjar använda detta läkemedel. Den innehåller information som är viktig för dig.
- Spara denna information, du kan behöva läsa den igen.
- Om du har ytterligare frågor vänd dig till läkare eller apotekspersonal.
- Detta läkemedel har ordinerats enbart åt dig. Ge det inte till andra. Det kan skada dem, även om de uppvisar sjukdomstecken som liknar dina.
- Om du får biverkningar, tala med läkare eller apotekspersonal. Detta gäller även eventuella biverkningar som inte nämns i denna information. Se avsnitt Eventuella biverkningar.
I denna bipacksedel finns information om följande
- Vad Cluvot är och vad det används för
- Vad du behöver veta innan du använder Cluvot
- Hur du använder Cluvot
- Eventuella biverkningar
- Hur Cluvot ska förvaras
- Förpackningens innehåll och övriga upplysningar
Vad produkten är och vad den används för
Vad är Cluvot?
Cluvot består av ett vitt pulver och en spädningsvätska som bereds till en färdig lösning. Lösningen ges som injektion i en ven.
Cluvot innehåller koagulationsfaktor XIII (FXIII) från human plasma (vätskedelen i blodet), och har en viktig hemostatisk funktion (stoppar blödning).
Vad används Cluvot för?
Cluvot används till vuxna och barn
- som förebyggande behandling vid medfödd faktor XIII brist och
- för behandling av blödning under operation av patienter med medfödd FXIII-brist.
Human koagulationsfaktor XIII som finns i Cluvot kan också vara godkänd för att behandla andra sjukdomar som inte nämns i denna produktinformation. Fråga läkare, apotekspersonal eller annan hälsovårdspersonal om du har ytterligare frågor och följ alltid deras instruktion.
Vad du behöver veta innan produkten används
Följande avsnitt innehåller information som din läkare ska beakta innan du får Cluvot.
Använd inte Cluvot:
- om du är allergisk mot den aktiva substansen eller något annat innehållsämne i detta läkemedel (anges i avsnitt Förpackningens innehåll och övriga upplysningar).
Informera din läkare om du har någon läkemedels- eller födoämnesallergi.
Varningar och försiktighet
Tala med läkare innan du använder Cluvot:
- om du tidigare har fått allergiska reaktioner av FXIII. Du bör ta antihistaminer och kortikosteroider i förebyggande syfte om din läkare råder dig att göra det.
- om en allergisk eller anafylaktisk reaktion inträffar (en allvarlig allergisk reaktion som medför stora svårigheter att andas eller yrsel). Användningen av Cluvot ska då genast avbrytas (injektionen/infusionen stoppas). Vid medicinsk chock ska medicinsk standardbehandling användas.
- om du tidigare haft trombos (blodpropp). Försiktighet ska iakttas på grund av FXIIIs förmåga att inverka på blodets koagulation (fibrinstabiliserande effekt).
En känd komplikation av behandlingen är bildning av inhibitorer (neutraliserande antikroppar) vilket innebär att behandlingen slutar att fungera. Om din blödning inte kan hållas under kontroll med hjälp av Cluvot ska du omedelbart rådgöra med din läkare. Du ska då övervakas noggrant för bildning av antikroppar.
Din läkare kommer noggrant att överväga nyttan med behandling av Cluvot jämfört med risken för ovanstående komplikationer.
Virussäkerhet
När läkemedel framställs ur humant blod eller plasma vidtas speciella åtgärder för att förhindra att infektioner överförs till patienter. Detta inkluderar:
- ett noggrant urval av blod- och plasmagivare för att säkerställa att personer med risk för att vara smittbärare utesluts.
- test av enskilda donationer och plasmapooler för tecken på virus/infektioner.
- steg för att inaktivera eller ta bort virus vid framställningen ur blod och plasma.
Trots detta kan risken för överföring av infektion inte helt uteslutas när läkemedel framställda ur humant blod eller plasma ges. Detta gäller även okända eller nya virus eller andra typer av infektioner.
De åtgärder som vidtas anses vara effektiva mot höljeförsedda virus såsom humant immunbristvirus (HIV), hepatit B- och hepatit C-virus samt mot de icke höljeförsedda virusen hepatit A och parvovirus B19.
Det rekommenderas starkt att din läkare registrerar produktnamn och tillverkningssatsnummer på läkemedlet (anges på kartongen) varje gång du får Cluvot.
Din läkare kan komma att föreslå vaccination mot hepatit A och B om du regelbundet/upprepat behandlas med läkemedel framställda från human plasma.
Andra läkemedel och Cluvot
- Tala om för läkare eller apotekspersonal om du tar, nyligen har tagit eller kan tänkas ta andra läkemedel.
- Det finns inga kända interaktioner mellan koncentrat som innehåller human koagulationsfaktor XIII och andra läkemedel.
- Cluvot ska inte blandas med andra läkemedel, lösningar eller spädningsvätskor än de som anges i avsnitt Förpackningens innehåll och övriga upplysningar och dessa ska administreras med en separat infusionslinje.
Graviditet, amning och fertilitet
- Om du är gravid eller ammar, tror att du kan vara gravid eller planerar att skaffa barn, ska du rådfråga läkare eller apotekspersonal innan du använder detta läkemedel.
- Begränsade data från användning av Cluvot hos gravida kvinnor tyder inte på några skadliga effekter under graviditeten eller på fostret/det nyfödda barnets hälsa. Därför kan Cluvot användas under graviditet när detta anses nödvändigt.
- Det finns inga data på utsöndring av Cluvot i bröstmjölk. Eftersom FXIII är en stor molekyl är utsöndring i bröstmjölk inte trolig och beroende på dess proteinegenskaper är absorption av intakta molekyler hos barnet också osannolikt. Cluvot kan därför användas under amning.
- Det finns inga tillgängliga data angående fertilitet.
Körförmåga och användning av maskiner
Inga studier på förmågan att framföra fordon eller använda maskiner har utförts.
Du är själv ansvarig för att bedöma om du är i kondition att framföra motorfordon eller utföra arbeten som kräver skärpt uppmärksamhet. En av faktorerna som kan påverka din förmåga i dessa avseenden är användning av läkemedel på grund av deras effekter och/eller biverkningar. Beskrivning av dessa effekter och biverkningar finns i andra avsnitt. Läs därför all information i denna bipacksedel för vägledning. Diskutera med din läkare eller apotekspersonal om du är osäker.
Cluvot innehåller natrium
Var uppmärksam på att Cluvot innehåller natrium. Detta bör beaktas om du ordinerats saltfattig kost. Cluvot innehåller 124,4-195,4 mg (5,41-8,50 mmol) natrium per dos (40 IU/kg kroppsvikt - för en normalvikt på 70 kg), om den rekommenderade dosen (2800 IU = 44,8 ml) ges.
Hur produkten används
- Cluvot ges vanligtvis av en läkare.
- Cluvot är endast avsett att ges som en injektion i en ven.
Dosering
Läkaren kommer att beräkna den dos du behöver och hur ofta Cluvot ska ges till dig med hänsyn till hur väl behandlingen fungerar.
För mer information se avsnittet ”Följande uppgifter är endast avsedda för hälso-och sjukvårdspersonal”.
Om du har använt för stor mängd av Cluvot
Inga fall av överdosering har rapporterats och det är heller inte förväntat eftersom sjukvårdspersonal ger läkemedlet.
Eventuella biverkningar
Liksom alla läkemedel kan detta läkemedel orsaka biverkningar, men alla användare behöver inte få dem.
Följande biverkningar har observerats i sällsynta fall (förekommer hos fler än 1 av 10 000 och färre än 1 av 1000 användare):
- Allergiska reaktioner såsom urtikaria (kliande svullnader i huden), hudutslag, blodtrycksfall (vilket kan få dig att känna dig yr eller svimfärdig) och svårighet att andas.
- Förhöjd kroppstemperatur
Följande biverkningar har observerats i mycket sällsynta fall (förekommer hos färre än 1 av 10 000 användare):
- Bildning av antikroppar mot FXIII.
Om allergiska reaktioner uppstår måste administreringen av Cluvot omedelbart avbrytas och lämplig behandling påbörjas. Vid medicinsk chock ska gällande medicinsk standardbehandling tillämpas.
Biverkningar hos barn och ungdomar
Biverkningar hos barn och ungdomar förväntas vara desamma som hos vuxna.
Rapportering av biverkningar
Om du får biverkningar, tala med läkare, apotekspersonal eller sjuksköterska. Detta gäller även biverkningar som inte nämns i denna information. Du kan också rapportera biverkningar direkt (se detaljer nedan). Genom att rapportera biverkningar kan du bidra till att öka informationen om läkemedlets säkerhet.
Finland
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Sverige
Läkemedelsverket
Box 26
751 03 Uppsala
Webbplats: www.lakemedelsverket.se
Hur produkten ska förvaras
- Förvaras i kylskåp (2°C - 8°C).
- Får ej frysas.
- Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
- Cluvot innehåller inga konserveringsmedel. Den färdigberedda lösningen ska ges omedelbart. Om lösningen inte används omedelbart ska förvaringstiden inte överstiga 4 timmar i rumstemperatur. Den färdigberedda lösningen ska inte kyl- eller frysförvaras.
- Förvara detta läkemedel utom syn- och räckhåll för barn.
- Används före utgångsdatum som anges på etiketten och kartongen.
Förpackningens innehåll och övriga upplysningar
Innehållsdeklaration
Den aktiva substansen är:
Human koagulationsfaktor XIII 250 IU respektive 1250 IU per injektionsflaska.
IU = IE (Internationella enheter)
Övriga innehållsämnen är:
Humant albumin, glukosmonohydrat, natriumklorid, natriumhydroxid (i små mängder för justering av pH).
Spädningsvätska: Vatten för injektionsvätskor.
Läkemedlets utseende och förpackningsstorlekar
Cluvot tillhandahålls som ett vitt pulver tillsammans med en spädningsvätska (vatten för
injektionsvätskor). Den färdigberedda lösningen ska vara färglös, klar till lätt opaliserande. Då den hålls mot ljuset ska den inte vara grumlig eller innehålla fällning/partiklar.
Förpackning:
En förpackning med 250 IU innehåller
- 1 injektionsflaska med pulver
- 1 injektionsflaska med 4 ml vatten för injektionsvätskor
- 1 överföringsset med filter 20/20 (Mix2Vial)
Tillbehör för administrering (innerkartong):
- 1 engångsspruta (5 ml)
- 1 venpunktionsset
- 2 alkoholtorkar
- 1 icke-sterilt plåster
En förpackning med 1250 IU innehåller
- 1 injektionsflaska med pulver
- 1 injektionsflaska med 20 ml vatten för injektionsvätskor
- 1 överföringsset med filter 20/20 (Mix2Vial)
Tillbehör för administrering (innerkartong):
- 1 engångsspruta (20 ml)
- 1 venpunktionsset
- 2 alkoholtorkar
- 1 icke-sterilt plåster
Innehavare av godkännande för försäljning och tillverkare
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg
Tyskland
Ytterligare upplysningar om detta läkemedel kan erhållas av ombudet för innehavaren av godkännande för försäljning:
CSL Behring AB
Box 712
182 17 Danderyd
Sverige
Denna bipacksedel ändrades senast 21.12.2018.
-------------------------------------------------------------------------
Direktiv för experterna inom hälsovården
Dosering
1 ml motsvarar cirka 62,5 IU och 100 IU motsvarar 1,6 ml.
Viktigt:
Dosen som administreras och doseringsintervallet bör alltid anpassas till den kliniska effekten i varje enskilt fall.
Dos
Doseringsregimen bör individanpassas med hänsyn till kroppsvikt, laboratorievärden och patientens kliniska tillstånd.
Doseringsschema för långtidsprofylax
Initial dos
- 40 internationella enheter (IU) per kg kroppsvikt
- Injektionshastigheten ska inte överskrida 4 ml per minut.
Efterföljande dosering
- Doseringen ska vägledas av den senaste lägsta aktivitetsnivå av faktor XIII, med administrering var 28:e dag (4 veckor) för att bibehålla en lägsta aktivitetsnivå av faktor XIII på ungefär 5-20 %.
- Justeringar av rekommenderad dosering med ±5 IU per kg ska baseras på lägsta aktivitetsnivå av faktor XIII så som visas i tabell 1 samt patientens kliniska tillstånd.
- Doseringsjusteringar ska baseras på en specifik analysmetod med hög känslighet som utförs för att bestämma FXIII-nivåerna. Ett exempel på dosjustering med hjälp av Berichrom® standardaktivitetstest beskrivs nedan i tabell 1.
Tabell 1: Doseringsjusteringar genom användning av Berichrom® aktivitetstest
| Lägsta aktivitetsnivå av faktor XIII (%) | Dosförändring |
| En lägsta nivå på <5 % | Öka med 5 enheter per kg |
| En lägsta nivå på 5-20 % | Ingen förändring |
| Två lägsta nivåer på >20 % | Minska med 5 enheter per kg |
| En lägsta nivå på >25 % | Minska med 5 enheter per kg |
Styrkan uttryckt i enheter har bestämts med hjälp av Berichrom® aktivitetstest, refererat till den nuvarande internationella standarden för human koagulationsfaktor XIII. Följaktligen motsvarar en enhet en internationell enhet.
Profylax innan operation
Efter patientens sista dos av långtidsprofylax om en operation är planerad:
- 21-28 dagar senare – administrera patientens hela profylaktiska dos direkt innan operation och ge nästkommande profylaktiska dos efter 28 dagar.
- 8-21 dagar senare – ytterligare en dos (hel eller partiell) kan administreras innan operation. Dosen ska vägledas av patientens FXIII-aktivitetsnivå och kliniska tillstånd och bör justeras i enlighet med halveringstiden för Cluvot.
- Inom 7 dagar efter sista dosen – eventuellt är ytterligare dosering inte nödvändig.
Justeringar av doseringen kan skilja från dessa rekommendationer och ska anpassas i varje enskilt fall utifrån FXIII-aktivitetsnivå och patientens kliniska tillstånd. Alla patienter bör noggrant observeras under och efter operationen.
Därför rekommenderas noggrann kontroll av ökningen i FXIII-aktivitet med en FXIII-analys. Vid större kirurgiskt ingrepp och allvarliga blödningar är syftet att uppnå nära normala värden (friska personer: 70-140%).
Pediatrisk population
Dosering och administreringssätt till barn och ungdomar baseras på kroppsvikt och är därför generellt baserat på samma riktlinjer som för vuxna. Dos och/eller administreringsfrekvens för varje individ ska alltid vägledas av klinisk effekt och FXIII-aktivitetsnivå.
Äldre
Dosering och administreringssätt till äldre (>65 år) har inte dokumenterats i kliniska studier.
Administreringsätt
Allmänna instruktioner
- Lösningen ska vara klar eller lätt opaliserande. Efter filtrering/fyllning av sprutan (se nedan) ska den färdigberedda lösningen inspekteras visuellt med avseende på partiklar och missfärgning innan administrering.
- Lösning som är synbart grumlig eller som innehåller fällning eller partiklar ska inte användas.
- Beredning och fyllning av sprutan ska ske under aseptiska förhållanden.
Beredning
Värm vätskan till rumstemperatur. Avlägsna plastlocket både från injektionsflaskan med pulver och injektionsflaskan med vätska. Behandla gummipropparnas ytor med antiseptisk lösning och låt dem torka innan Mix2Vial-förpackningen öppnas.
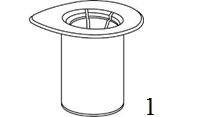 |
1. Öppna Mix2Vial-förpackningen genom att dra av förslutningen. Ta inte ut Mix2Vial-setet ur blisterförpackningen! |
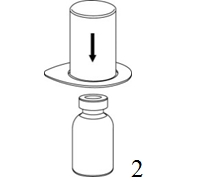 |
2. Placera injektionsflaskan med vätska på en plan, ren yta och håll fast injektionsflaskan stadigt. Ta blisterförpackningen med Mix2Vial-setet och tryck spetsen på den blå adapterdelen rakt ner genom proppen på injektionsflaskan med vätska. |
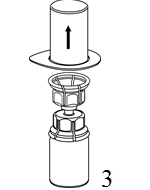 |
3. Ta försiktigt bort blisterförpackningen från Mix2Vial-setet genom att ovanifrån ta tag om kanten och dra rakt upp. Se till att endast blisterförpackningen tas bort, inte Mix2Vial-setet. |
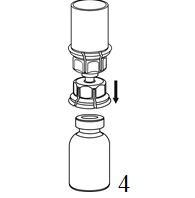 |
4. Ställ injektionsflaskan med pulver på ett plant och fast underlag. Vänd injektionsflaskan med vätska med det fastsatta Mix2Vial-setet och tryck spetsen på den transparenta adapterdelen rakt ner genom proppen på injektionsflaskan med pulver. Vätskan kommer automatiskt att rinna över till injektionsflaskan med pulver. |
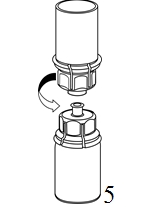 |
5. Fatta tag i den del av Mix2Vial-setet där injektionsflaskan med pulver sitter med ena handen och den del av Mix2Vial-setet där injektionsflaskan med vätska sitter med den andra handen och skruva försiktigt isär setet i två delar. Kassera injektionsflaskan med vätska med den blå Mix2Vial-delen fastsatt. |
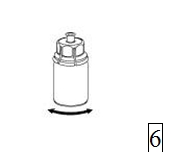 |
6. Rotera försiktigt injektionsflaskan med pulver med den fastsatta transparenta adaptern tills pulvret löst sig fullständigt. Skaka inte injektionsflaskan. |
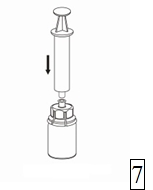 |
7. Dra in luft i en tom, steril spruta. Medan injektionsflaskan med pulver står rakt upp kopplas sprutan ihop med Luer-lock inpassningen på Mix2Vial-delen. |
Spruta in luft i injektionsflaskan.
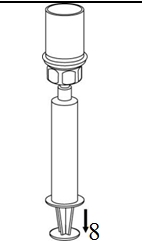 |
8. Med sprutkolven intryckt, vänds injektionsflaskan upp och ned tillsammans med set och spruta. Dra in lösningen i sprutan genom att långsamt föra kolven tillbaka. |
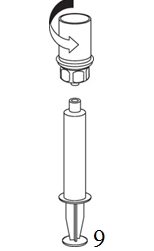 |
9. När all lösning har förts över till sprutan, fatta ett fast tag om sprutan (håll sprutan med kolven nedåt) och koppla bort den transparenta Mix2Vial-adaptern från sprutan. |
Var noggrann med att se till att inget blod kommer in i den fyllda sprutan eftersom det finns en risk för att blodet kan koagulera i sprutan och fibrinkoagel administreras till patienten.
Beredningen ska administreras i en separat injektions-/infusionslinje (medföljer produkten) genom långsam intravenös injektion/infusion med en hastighet som inte får överstiga 4 ml per minut.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.