COFACT pulver och vätska till injektionsvätska, lösning 250 IU, 500 IU
Tilläggsinformation
Cofact 250 IU pulver och vätska till injektionsvätska, lösning
Cofact 500 IU pulver och vätska till injektionsvätska, lösning
protrombinkomplex, humant
Allmänna direktiv
Läs noga igenom denna bipacksedel innan du börjar använda detta läkemedel. Den innehåller information som är viktig för dig.
- Spara denna information, du kan behöva läsa den igen.
- Om du har ytterligare frågor, vänd dig till läkare eller apotekspersonal.
- Detta läkemedel har ordinerats enbart åt dig. Ge det inte till andra. Det kan skada dem, även om de uppvisar sjukdomstecken som liknar dina.
- Om du får biverkningar, tala med läkare eller apotekspersonal. Detta gäller även eventuella biverkningar som inte nämns i denna information. Se avsnitt Eventuella biverkningar.
I denna bipacksedel finns information om följande
- Vad Cofact är och vad det används för
- Vad du behöver veta innan du använder Cofact
- Hur du använder Cofact
- Eventuella biverkningar
- Hur Cofact ska förvaras
- Förpackningens innehåll och övriga upplysningar
Vad produkten är och vad den används för
Cofact innehåller de humana koagulationsfaktorerna II, VII, IX och X som aktiva substanser.
Dessa koagulationsfaktorer är normala beståndsdelar i människans blod och kallas allmänt för protrombinkomplexet. De är vitamin K-beroende. Brist på en eller flera av dessa koagulationsfaktorer kan ge upphov till koagulationsstörningar i blodet. Som följd av detta ökar benägenheten för blödningar. Cofact ersätter koagulationsfaktorbristen och behandlar eller förebygger därmed blödningar.
Cofact kan användas för:
Behandling eller förebyggande av blödningar som orsakas av
- förvärvad brist på koagulationsfaktorer i protrombinkomplexet. Exempel på sådana är bristtillstånd som orsakats av K‑vitaminantagonistbehandling eller K-vitaminantagonistöverdos, varmed omedelbar korrigering av bristtillståndet krävs.
- medfödd brist på K-vitaminberoende koagulationsfaktorer då renade och specifika koagulationsfaktorer inte finns till förfogande.
Vad du behöver veta innan produkten används
Använd inte Cofact
- Om du är allergisk mot aktiva substanser eller något annat innehållsämne i detta läkemedel (anges i avsnitt Förpackningens innehåll och övriga upplysningar).
Varningar och försiktighet
Tala med läkare som är specialiserad på behandling av koagulationsstörningar innan du får Cofact.
- Om du har förvärvad brist på de vitamin K-beroende koagulationsfaktorerna (till exempel på grund av att du behandlats med läkemedel som hämmar effekten av vitamin K), får Cofact användas endast när en snabb korrigering av protrombinkomplexnivåerna är nödvändigt, t.ex. vid större blödning eller vid akut kirurgi. I andra fall räcker det vanligtvis med att minska dosen av vitamin K-antagonisten och/eller att ge vitamin K.
- Om du genomgår behandling med en vitamin K-antagonist kan du löpa ökad risk för blodpropp. I sådana fall kan behandling med Cofact öka denna risk.
- Om du har medfödd brist på någon vitamin K-beroende koagulationsfaktor: I dessa fall ska specifika koagulationsfaktorprodukter användas när sådana finns att tillgå.
- Om du får en allergisk eller anafylaxiliknande reaktion ska infusionen med Cofact genast avbrytas.
Det finns en risk för blodproppar eller disseminerad intravaskulär koagulation (ett tillstånd där blodet börjar koagulera i blodkärlen) när du får Cofact, särskilt om du får det upprepade gånger.
- Läkaren kontrollerar om det vid användningen av Cofact finns risk för blodproppar, se avsnitt Eventuella biverkningar Eventuella biverkningar). Risken för blodproppar är förhöjd för följande personer:
- personer som har haft en hjärtinfarkt eller som har eller har haft andra kranskärlssjukdomar
- personer med leversjukdom
- nyfödda
- personer som nyligen blivit opererade
- personer som löper ökad risk för koagulationskomplikationer (t.ex. personer som tidigare har haft tromboemboliska händelser eller disseminerad intravaskulär koagulation).
Din läkare kommer att noggrant överväga nyttan med behandling med Cofact jämfört med risken för dessa komplikationer.
Virussäkerhet
Då produkter framställs av humanblod eller -plasma vidtas särskilda åtgärder för att förhindra att infektioner överförs till patienter. Detta innebär
- ett noggrant urval av blod- och plasmadonatorer för att säkerställa att donatorer med risk för att bära på en infektion utesluts
- att varje donation och plasmapool testas för tecken på virus/infektion.
- införandet av steg i hanteringen av blod och plasma som kan inaktivera eller avskilja virus.
Trots dessa åtgärder kan smittorisken inte fullständigt uteslutas när läkemedel framställda av humanblod eller -plasma ges. Detta gäller också hittills okända eller nya virus eller andra typer av infektioner.
De åtgärder som vidtagits anses vara effektiva på höljeförsedda virus som t.ex. humant immunbristvirus (HIV), hepatit B-virus (HBV) och hepatit C-virus (HCV) samt på det icke-höljeförsedda viruset hepatit A (HAV). Åtgärderna kan vara av begränsat värde mot icke-höljeförsedda virus såsom parvovirus B19. Parvovirus B19-infektion kan vara allvarligt för gravida kvinnor (infektion av fostret) samt för personer med försvagat immunsystem eller med anemi (t.ex. sicklecellanemi eller hemolytisk anemi).
Särskilt rekommenderas det att produktens namn och satsnummer registreras varje gång du får en dos Cofact för att kunna upprätthålla ett register på använda produktsatser.
Barn och ungdomar
Det finns inga data angående användning av Cofact hos barn eller ungdomar.
Andra läkemedel och Cofact
Tala om för läkare eller apotekspersonal om du använder, nyligen har använt eller kan tänkas använda andra läkemedel, även receptfria sådana.
Det finns ingen information tillgänglig om eventuella samverkningar mellan Cofact och andra mediciner, med undantag av bloduttunnande mediciner.
Graviditet och amning
Om du är gravid eller ammar, tror att du kan vara gravid eller planerar att skaffa barn, rådfråga läkare innan du använder detta läkemedel. Om du är gravid eller ammar kommer du att ges Cofact endast om det är klart nödvändigt.
Körförmåga och användning av maskiner
Inga studier på förmågan att framföra eller använda maskiner har utförts.
Cofact innehåller natrium
Cofact innehåller upp till 448 mg natrium (huvudingrediensen i koksalt/bordssalt) per 100 ml. Detta motsvarar 22 % av högsta rekommenderat dagligt intag av natrium för vuxna. Detta bör beaktas om du ordinerats saltfattig kost.
Hur produkten används
Behandlingen ska påbörjas, ges och övervakas av en läkare med erfarenhet av behandling av koagulationsstörningar. Läkaren ordinerar den nödvändiga Cofact-dosen för behandling eller förebyggande av blödningar som en följd av användandet av bloduttunnande mediciner eller för behandling av en medfödd brist på K-vitaminberoende koagulationsfaktorer.
Den exakta dosen beror på
- hur allvarligt ditt tillstånd är
- din vikt
- de koagulationsfaktorer som du behöver
- koncentrationerna av dessa faktorer i ditt blod.
Vid medfödd koagulationsfaktorbrist är det viktigt att koagulationsfaktorernas blodkoncentrationer regelbundet fastställs.
Information för hälso- och sjukvårdspersonal finns i slutet av denna bipacksedel.
Om du använt för stor mängd av Cofact
Din läkare ska regelbundet kontrollera ditt blods koagulationsförmåga under behandlingen. Höga doser av protrombinkomplexkoncentrat har förknippats med fall av hjärtattack, disseminerad intravaskulär koagulation och en ökad bildning av blodproppar i blodkärlen hos patienter som har en ökad risk för dessa komplikationer.
Eventuella biverkningar
Liksom alla läkemedel kan detta läkemedel orsaka biverkningar, men alla användare behöver inte få dem.
Följande biverkningar har observerats:
Vanliga (kan förekomma hos upp till 1 av 10 användare):
- Det finns en risk för att blodproppar uppkommer (se avsnitt Vad du behöver veta innan produkten används).
Mindre vanliga (kan förekomma hos upp till 1 av 100 användare):
- Det finns en risk för att blodtrycket sjunker.
Följande biverkningar har rapporterats (frekvens kan inte beräknas från tillgängliga data):
- Överkänslighet eller allergiska reaktioner (se avsnitt Vad du behöver veta innan produkten används)
- Hjärtinfarkt
- Illamående, kräkningar
- Rodnad vid infusionsstället, irritation vid infusionsstället, svullnad vid infusionsstället, sjukdomskänsla
- Tillfällig ökning av leverfunktionsvärden
- Stroke, yrsel
- Lungemboli, andningssvårigheter
- Överdriven svettning, klåda i huden, nässelutslag, hudutslag
Hos patienter med brist på koagulationsfaktorerna II, VII, IX eller X, kan det som en följd av Cofact-behandlingen utvecklas antikroppar mot faktorerna i fråga. Då har läkemedlet inte önskad effekt.
Rapportering av biverkningar
Om du får biverkningar, tala med läkare eller apotekspersonal. Detta gäller även biverkningar som inte nämns i denna information. Du kan också rapportera biverkningar direkt (se detaljer nedan). Genom att rapportera biverkningar kan du bidra till att öka informationen om läkemedels säkerhet.
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Hur produkten ska förvaras
Förvara detta läkemedel utom syn- och räckhåll för barn.
Används före utgångsdatum som anges på förpackningen efter EXP. Utgångsdatumet är den sista dagen i angiven månad.
Förvaras i kylskåp (2oC-8oC). Får ej frysas.
Förvara injektionsflaskan i ytterförpackningen. Ljuskänsligt.
Cofact kan förvaras vid högst 25 °C i upp till sex månader, t.ex. under resor, utan att effekten försämras. Datum då produkten tagits i rumstemperatur ska antecknas på förpackningen. Om produkten inte används inom sex månader efter att den tagits i rumstemperatur måste den kasseras.
Efter att produkten har tagits ut ur kylskåp får den inte ställas tillbaka i kylskåp.
Hållbarhet för upplöst produkt har visats i högst 3 timmar vid 15 °C–25 °C. För att undvika kontaminering ska den upplösta produkten dock användas omedelbart.
Läkemedel ska inte kastas i avloppet eller bland hushållsavfall. Fråga apotekspersonalen hur man kastar läkemedel som inte längre används. Dessa åtgärder är till för att skydda miljön.
Förpackningens innehåll och övriga upplysningar
Innehållsdeklaration
- De aktiva substanserna är koagulationsfaktorerna II, VII, IX och X och ytterligare aktiva substanser är protein C och protein S.
- En injektionsflaska Cofact 250 IU innehåller 250 IU av faktor IX; 140 - 350 IU av faktor II; 70 - 200 IU av faktor VII och 140 - 350 IU av faktor X; 111–390 IU protein C; 10–80 IU protein S.
- En injektionsflaska Cofact 500 IU innehåller 500 IU av faktor IX; 280 - 700 IU av faktor II; 140 - 400 IU av faktor VII och 280 - 700 IU av faktor X; 222–780 IU protein C; 20–160 IU protein S.
När injektionspulvret har lösts upp i vattnet för injektionsvätskor som finns i förpackningen innehåller den färdigberedda lösningen:
- 14 - 35 IU av faktor II per ml
- 7 - 20 IU av faktor VII per ml
- 25 IU av faktor IX per ml
- 14 - 35 IU av faktor X per ml
- 11–39 IU protein C per ml
- 1–8 IU protein S per ml
- Övriga innehållsämnen är natriumcitrat, natriumklorid och antitrombin.
Läkemedlets utseende och förpackningsstorlekar
Cofact pulver till injektionsvätska är ett blåaktigt pulver. Den färdigberedda injektionslösningen är en blåaktig lösning.
Cofact tillhandahålls som pulver och vätska till injektionsvätska, lösning.
Innehåll i förpackningen med 250 IU
1 injektionsflaska med 250 IU pulver
1 injektionsflaska med 10 ml vatten för injektionsvätskor
1 överföringskanyl
Innehåll i förpackningen med 500 IU
1 injektionsflaska med 500 IU pulver
1 injektionsflaska med 20 ml vatten för injektionsvätskor
1 överföringskanyl
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Innehavare av godkännandet för försäljning och tillverkare
Prothya Biosolutions Netherlands B.V.
Plesmanlaan 125
NL-1066 CX Amsterdam
Nederländerna
Detta läkemedel är godkänt inom Europeiska ekonomiska samarbetsområdet under namnen:
Österrike, Belgien, Finland, Frankrike, Tyskland, Island, Italien, Luxemburg, Nederländerna och Spanien: Cofact
Sverige: Thyaplex
Denna bipacksedel ändrades senast 22.11.2022
Direktiv för experterna inom hälsovården
Följande uppgifter är endast avsedda för hälso- och sjukvårdspersonal:
Kvalitativ och kvantitativ sammansättning
Cofact (koncentrat av fyra koagulationsfaktorer) tillhandahålls som ett pulver och vätska till injektionsvätska, lösning och innehåller humant protrombinkomplex. Läkemedlet innehåller nominellt följande mängd IU av humana koagulationsfaktorer:
|
|
Cofact 250 IU (faktor IX) |
Cofact 500 IU (faktor IX) |
Efter beredning* |
|
Aktiva substanser |
|
|
|
|
Koagulationsfaktor II |
140–350 |
280–700 |
14–35 |
|
Koagulationsfaktor VII |
70–200 |
140–400 |
7–20 |
|
Koagulationsfaktor IX |
250 |
500 |
25 |
|
Koagulationsfaktor X |
140–350 |
280–700 |
14–35 |
|
Övriga aktiva substanser |
|
|
|
|
Protein C |
111–390 |
222–780 |
11–39 |
|
Protein S |
10–80 |
20–160 |
1–8 |
|
*Efter beredning med 10 ml (för Cofact 250 IU) eller 20 ml (för Cofact 500 IU) vatten för injektionsvätskor. |
|||
Det totala proteininnehållet per injektionsflaska är 130–350 mg (Cofact 250 IU) eller 260–700 mg (Cofact 500 IU). Produktens specifika aktivitet är ≥ 0,6 IU/mg, utryckt som faktor IX-aktivitet.
Aktiviteterna hos samtliga koagulationsfaktorer såväl som hos protein C och S (antigen) har testats enligt gällande standarder för WHO eller Europafarmakopén.
Efter beredning innehåller detta läkemedel 125–195 mmol natrium/l, upp till 44,8 mg natrium per 10 ml.
Dosering och administreringssätt
Dosering
Endast allmänna riktlinjer för dosering ges nedan. Behandling bör sättas in under överinseende av läkare med erfarenhet av behandling av koagulationsrubbningar. Substitutionsbehandlingens dosering och duration avgörs av rubbningens svårighetsgrad, blödningens lokalisation och omfattning samt patientens kliniska tillstånd.
Dos och doseringsintervall ska beräknas individuellt för varje patient. Doseringsintervallet måste anpassas till att olika koagulationsfaktorer i protrombinkomplexet har olika halveringstid i plasma. Individuella doseringsbehov kan bara bestämmas utifrån regelbundna mätningar av individuella plasmanivåer för koagulationsfaktorn i fråga, eller genom globala test på protrombinkomplexnivåerna (PK, INR) och kontinuerlig övervakning av patientens kliniska tillstånd.
Vid större kirurgiska ingrepp är noggrann övervakning av substitutionsbehandlingen med hjälp av koagulationsanalyser nödvändig (specifika koagulationsfaktoranalyser och/eller globala test på protrombinkomplexnivåerna).
Blödning och perioperativ blödningsprofylax under behandling med vitamin K-antagonist:
Dosen avgörs av INR före behandling, målvärdet för INR och kroppsvikten. I nedanstående tabell anges de ungefärliga doser som krävs för korrigering av INR vid olika initiala INR-nivåer.
Doseringstabellerna ska endast användas som allmänna riktlinjer och de kan inte ersätta den individuella doseringsberäkningen för varje enskild patient samt en noggrann övervakning av INR och andra koagulationsparametrar vid behandling.
Rekommenderade doser av Cofact i ml för att uppnå målvärdet för INR på ≤ 2,1
|
Initialt INR-värde |
7,5 |
5,9 |
4,8 |
4,2 |
3,6 |
3,3 |
3,0 |
2,8 |
2,6 |
2,5 |
2,3 |
2,2 |
| Kroppsvikt | ||||||||||||
|
50 kg |
40 |
40 |
40 |
30 |
30 |
30 |
20 |
20 |
X |
X |
X |
X |
|
60 kg |
50 |
50 |
40 |
40 |
30 |
30 |
30 |
20 |
X |
X |
X |
X |
|
70 kg |
60 |
50 |
50 |
50 |
40 |
40 |
30 |
30 |
X |
X |
X |
X |
|
80 kg |
60 |
60 |
60 |
50 |
50 |
40 |
40 |
30 |
X |
X |
X |
X |
|
90 kg |
60 |
60 |
60 |
60 |
50 |
50 |
40 |
30 |
X |
X |
X |
X |
|
100 kg |
60 |
60 |
60 |
60 |
60 |
50 |
40 |
40 |
X |
X |
X |
X |
Rekommenderade doser av Cofact i ml för att uppnå målvärdet för INR på ≤ 1,5
|
Initialt INR-värde |
7,5 |
5,9 |
4,8 |
4,2 |
3,6 |
3,3 |
3,0 |
2,8 |
2,6 |
2,5 |
2,3 |
2,2 |
| Kroppsvikt | ||||||||||||
|
50 kg |
60 |
60 |
60 |
50 |
50 |
50 |
40 |
40 |
30 |
30 |
30 |
30 |
|
60 kg |
80 |
70 |
70 |
60 |
60 |
60 |
50 |
50 |
40 |
40 |
40 |
30 |
|
70 kg |
90 |
80 |
80 |
70 |
70 |
70 |
60 |
60 |
50 |
40 |
40 |
40 |
|
80 kg |
100 |
100 |
90 |
90 |
90 |
80 |
80 |
70 |
60 |
50 |
50 |
40 |
|
90 kg |
100 |
100 |
100 |
90 |
90 |
90 |
80 |
80 |
70 |
60 |
50 |
40 |
|
100 kg |
100 |
100 |
100 |
100 |
100 |
90 |
90 |
80 |
70 |
70 |
60 |
50 |
Doserna beräknas baserat på koncentrationen av faktor IX i Cofact, eftersom den har relativt kort halveringstid och lågt utbyte efter infusion jämfört med övriga koagulationsfaktorer i protrombinkomplexet. Det antas att en genomsnittlig plasmakoncentration av faktor IX på ≥ 30 % är tillräcklig för att uppnå ett INR-värde på ≤ 2,1 och ≥ 60 % för att uppnå ett INR-värde på ≤ 1,5. Beräknade mängder avrundas till multiplar om 10 ml och en övre gräns på totalt 60 eller 100 ml har fastställts (se tabellerna ovan). Målvärdena för INR rekommenderas av Federation of Dutch Thrombosis Services och de är liknande som rekommendationerna i England och Tyskland.
Justeringen av en vitamin K-antagonistinducerad rubbning av hemostasen kvarstår i ungefär 6–8 timmar. Dock inträder effekterna av vitamin K, om detta givits samtidigt, vanligtvis inom 4–6 timmar. Därför krävs vanligen inte upprepad behandling med humant protrombinkomplexkoncentrat när vitamin K har givits.
Då dessa rekommendationer är empiriskt grundade, och recovery och effektduration kan variera, krävs övervakning av behandlingen med INR-bestämningar.
Blödning och perioperativ profylax vid medfödd brist på någon av de vitamin K-beroende koagulationsfaktorerna, när specifika koagulationsfaktorprodukter inte finns att tillgå:
Den beräknade dosering som krävs för behandling baseras på det empiriska fyndet att ungefär 1 IU av faktor VII eller faktor IX per kg kroppsvikt höjer plasmaaktiviteten för faktor VII respektive IX med 0,01 IU/ml, medan 1 IU av faktor II eller X per kg kroppsvikt höjer plasmaaktiviteten för faktor II eller X med 0,02 respektive 0,017 IU/ml.
Dosen för en specifik faktor uttrycks i internationella enheter (IU), vilka relateras till den aktuella WHO-standarden för varje faktor. Aktiviteten i plasma för en specifik koagulationsfaktor uttrycks antingen som en procentandel (i förhållande till normal plasma) eller i internationella enheter (i förhållande till den internationella standarden för den specifika koagulationsfaktorn).
En internationell enhet (IU) koagulationsfaktoraktivitet motsvarar mängden i 1 ml normal humanplasma.
Exempelvis baseras beräkningen av den erforderliga doseringen av faktor X på det empiriska fyndet att 1 internationell enhet (IU) av faktor X per kg kroppsvikt höjer aktiviteten av faktor X i plasma med 0,017 IU/ml. Den erforderliga dosen beräknas med hjälp av följande formel:
Erforderligt antal enheter = kroppsvikt (kg) x önskad ökning av faktor X (IU/ml) x 60
Där 60 (ml/kg) är det reciproka värdet för uppskattad recovery.
Om individuell recovery är känd ska detta värde användas vid beräkning.
Administreringssätt
För anvisningar om beredning av läkemedlet före administrering, se avsnitt Särskilda anvisningar för destruktion och övrig hantering. Cofact ska administreras intravenöst.
Det rekommenderas att den färdigberedda produkten administreras med en hastighet av ungefär 2 ml/minut.
Pediatrisk population
Säkerhet och effekt för Cofact för pediatriska patienter har inte fastställts.
Inkompatibiliteter
Detta läkemedel får inte blandas med andra läkemedel.
Cofact är kompatibelt med polypropenmaterial. Behandlingsfel kan inträffa till följd av adsorption av koagulationsfaktorn till de inre ytorna av annan injektions-/infusionsutrustning.
Hållbarhet
3 år.
Kemisk och fysikalisk stabilitet efter beredning har visats i 3 timmar vid 15 °C–25 °C. Ur mikrobiologiskt perspektiv ska produkten användas omedelbart efter beredning. Om den inte används omedelbart är förvaringstiden och förhållandena före användning användarens ansvar.
Särskilda anvisningar för destruktion och övrig hantering
Upplösning
Den torkade proteinfraktionen ska lösas i den föreskrivna volymen vatten för injektionsvätskor. Vid förvaring vid 2 °C–8 °C måste injektionsflaskorna med Cofact och vatten för injektionsvätskor ha nått rumstemperatur (15 °C–25 °C) innan upplösningen genomförs.
Procedur med hjälp av en överföringskanyl
- Ta bort det skyddande plastlocket från flaskan som innehåller vatten för injektionsvätskor och flaskan som innehåller produkten.
- Desinficera flaskornas gummiproppar med en kompress fuktad med alkohol (70 %).
- Ta bort det skyddande locket från den ena änden av överföringskanylen och stick in kanylen i flaskan som innehåller vatten för injektionsvätskor (A).
- Ta sedan bort det skyddande locket från den andra änden av överföringskanylen, vänd flaskan med överföringskanylen upp och ned och stick omedelbart in den fria kanylen i flaskan som innehåller produkten (B).
Undertrycket i den flaska som innehåller produkten kommer att göra att vattnet för injektionsvätskor sugs in i flaskan. Rekommendation: när vattnet för injektionsvätskor rinner mellan flaskorna, ska den flaska som innehåller produkten hållas lutad så att vattnet rinner utmed flaskans vägg. Det gör att produkten löser upp sig snabbare. Så snart vattnet har runnit över i den andra flaskan ska den tomma flaskan och överföringskanylen tas bort i en enda rörelse.
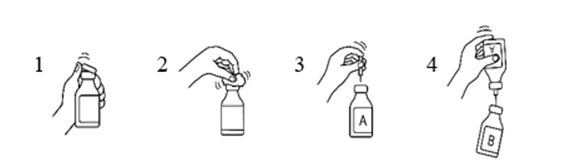
För att påskynda upplösningen kan man snurra flaskan försiktigt och, om det är nödvändigt, värma den till 30 °C. Flaskan får inte skakas och temperaturen får inte överstiga 37 °C. Om flaskan värms i vattenbad, ska försiktighet iakttas så att vattnet inte kommer i kontakt med det skyddande locket och/eller gummiproppen.
Den torkade substansen ska i regel ha löst upp sig fullständigt inom 10 minuter och bildat en blåfärgad lösning. Den blå färgen orsakas av plasmaproteinet ceruloplasmin.
Lösningen ska vara klar eller något opaliserande. Använd inte lösningar som är grumliga eller som innehåller fällning. Färdigberedd produkt ska granskas visuellt med avseende på partiklar och missfärgning före administrering.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.