COFACT injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU
Cofact 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Cofact 500 IU injektiokuiva-aine ja liuotin, liuosta varten
ihmisen protrombiinikompleksi
Yleisiä ohjeita
Lue tämä pakkausseloste huolellisesti ennen kuin aloitat lääkkeen käyttämisen, sillä se sisältää sinulle tärkeitä tietoja.
- Säilytä tämä pakkausseloste. Voit tarvita sitä myöhemmin.
- Jos sinulla on kysyttävää, käänny lääkärin tai apteekkihenkilökunnan puoleen.
- Tämä lääke on määrätty vain sinulle eikä sitä tule antaa muiden käyttöön. Se voi aiheuttaa haittaa muille, vaikka heillä olisikin samanlaiset oireet kuin sinulla.
- Jos havaitset haittavaikutuksia, käänny lääkärin tai apteekkihenkilökunnan puoleen. Tämä koskee myös sellaisia mahdollisia haittavaikutuksia, joita ei ole mainittu tässä pakkausselosteessa. Ks. kohta Mahdolliset haittavaikutukset.
Tässä pakkausselosteessa kerrotaan
- Mitä Cofact on ja mihin sitä käytetään
- Mitä sinun on tiedettävä, ennen kuin käytät Cofactia
- Miten Cofactia käytetään
- Mahdolliset haittavaikutukset
- Cofactin säilyttäminen
- Pakkauksen sisältö ja muuta tietoa
Mitä valmiste on ja mihin sitä käytetään
Cofact sisältää vaikuttavina aineina hyytymistekijöitä II, VII, IX ja X, jotka ovat ihmisen veren hyytymistekijöitä.
Nämä hyytymistekijät ovat ihmisen veren normaaleja aineosia, ja niitä kutsutaan yleisesti protrombiinikompleksiksi. Ne ovat K-vitamiiniriippuvaisia. Yhden tai useamman hyytymistekijän puutos voi aiheuttaa veren hyytymishäiriöitä. Niiden seurauksena alttius verenvuodoille voi lisääntyä. Cofact korvaa puuttuvia hyytymistekijöitä ja tyrehdyttää tai ehkäisee verenvuotoja.
Cofactia voidaan käyttää seuraavissa tapauksissa:
Sellaisten verenvuotojen hoito tai ehkäiseminen, jotka aiheutuvat
- hankinnaisesta protrombiinikompleksin hyytymistekijöiden puutoksesta. Esimerkkejä tällaisista ovat K-vitamiiniantagonistihoidon tai K-vitamiiniantagonistien yliannostuksen aiheuttamat puutostilat, jolloin tarvitaan välitöntä puutoksen korjaamista.
- synnynnäisestä K-vitamiinista riippuvien hyytymistekijöiden puutoksesta, jos puhdistettuja ja spesifisiä hyytymistekijöitä ei ole saatavilla.
Mitä sinun on tiedettävä ennen valmisteen käyttöä
Älä käytä Cofactia
- jos olet allerginen vaikuttaville aineille tai tämän lääkkeen jollekin muulle aineelle (lueteltu kohdassa Pakkauksen sisältö ja muuta tietoa).
Varoitukset ja varotoimet
Keskustele ennen Cofact-valmisteen saamista hyytymishäiriöiden hoitoon erikoistuneen lääkärin kanssa
- jos sinulla on hankinnainen K-vitamiiniriippuvaisten hyytymistekijöiden puutos (esimerkiksi K‑vitamiinin estäjälääkityksen vuoksi), Cofact-valmistetta saa käyttää vain, jos protrombiinikompleksipitoisuuden nopea korjaaminen on tarpeen esim. vakavien verenvuotojen tai päivystysluonteisen leikkauksen yhteydessä. Muissa tapauksissa riittävät K-vitamiinin estäjän annoksen pienentäminen ja/tai K-vitamiinin anto.
- jos saat K-vitamiinin estäjälääkitystä, sinulla voi olla tavanomaista suurempi veritulppien muodostumisen riski; Cofact-hoito voi tällöin lisätä tätä riskiä
- jos sinulla on synnynnäinen jonkin K-vitamiiniriippuvaisen hyytymistekijän puutos (perinnöllinen puutos), on käytettävä kyseistä hyytymistekijää sisältävää valmistetta, jos sellaista on saatavilla
- jos ilmenee allerginen tai anafylaktistyyppinen reaktio, Cofact-infuusio pitää lopettaa välittömästi.
Cofact-valmistetta saadessasi on olemassa verisuonitukoksen tai disseminoituneen intravaskulaarisen koagulaation riski (eli veritulppien muodostuminen verisuonissa), etenkin jos saat Cofact-valmistetta toistuvasti.
- Hoitava lääkäri arvioi, sisältyykö Cofact-valmisteen antamiseen verisuonitukoksen riski (ks. kohta Mahdolliset haittavaikutukset Mahdolliset haittavaikutukset). Verisuonitukosten riski on suurentunut seuraavilla henkilöillä:
- henkilöillä, joilla on ollut sydäninfarkti tai joilla on ollut (tai on edelleen) muita sepelvaltimosairauksia
- henkilöillä, joilla on maksasairaus
- vastasyntyneillä
- henkilöillä, jotka ovat äskettäin olleet leikkauksessa
- henkilöillä, joilla veren hyytymiseen liittyvät komplikaatiot ovat todennäköisempiä (esim. joilla on aiemmin ollut tromboembolisia tapahtumia tai disseminoitunut intravaskulaarinen koagulaatio).
Lääkäri arvioi tarkoin Cofact-hoidon hyödyn näiden komplikaatioiden riskiin nähden.
Virusturvallisuus
Kun lääkkeitä valmistetaan ihmisen verestä tai plasmasta, käytetään tiettyjä toimenpiteitä, joilla estetään infektioiden siirtyminen potilaisiin. Näitä toimenpiteitä ovat
- veren ja plasman luovuttajien huolellinen valinta, jotta voidaan sulkea pois luovutuksista mahdolliset infektioiden kantajat
- jokaisen luovutuksen ja plasmapoolin tutkiminen virusten/infektioiden tunnusmerkkien suhteen
- sellaisten vaiheiden lisääminen valmistukseen, jotka tekevät viruksia tehottomiksi tai poistavat viruksia.
Näistä toimenpiteistä huolimatta infektion siirtymismahdollisuutta ei voida sulkea täysin pois, kun annetaan ihmisen verestä tai plasmasta valmistettuja lääkkeitä. Tämä koskee myös tuntemattomia tai uusia viruksia tai muun tyyppisiä infektioita.
Tehtyjen toimenpiteiden voidaan katsoa olevan tehokkaita vaipallisiin viruksiin, kuten ihmisen immuunikatovirus (HIV), hepatiitti B ‑virus (HBV) ja hepatiitti C ‑virus (HCV), sekä vaipattomaan hepatiitti A ‑virukseen (HAV). Tehokkuus muihin vaipattomiin viruksiin, kuten parvovirus B19, saattaa olla rajallinen. Parvovirus B19 ‑tartunta saattaa olla vakava raskaana oleville naisille (sikiön saama tartunta) sekä henkilöille, joiden immuunijärjestelmä on heikentynyt tai joilla on anemia (esim. sirppisoluanemia tai hemolyyttinen anemia).
Erityisesti suositellaan, että joka kerta, kun saat Cofactia, tuotteen nimi ja eränumero kirjataan ylös, jotta voidaan pitää yllä tiedostoa käytetyistä eristä.
Lapset ja nuoret
Cofact-valmisteen käytöstä lapsille ja nuorille ei ole tietoja saatavissa.
Muut lääkevalmisteet ja Cofact
Kerro lääkärille tai apteekkihenkilökunnalle, jos parhaillaan käytät, olet äskettäin käyttänyt tai saatat käyttää muita lääkkeitä, myös lääkkeitä, joita lääkäri ei ole määrännyt.
Cofactin ja muiden lääkkeiden, lukuun ottamatta verenohennuslääkkeitä, mahdollisista yhteisvaikutuksista ei ole tietoja.
Raskaus ja imetys
Jos olet raskaana tai imetät, epäilet olevasi raskaana tai jos suunnittelet lapsen hankkimista, kysy lääkäriltä neuvoa ennen tämän lääkkeen käyttöä. Jos olet raskaana tai imetät, lääkäri antaa sinulle Cofact-valmistetta vain, jos se on selkeästi aiheellista.
Ajaminen ja koneiden käyttö
Vaikutuksia ajokykyyn tai koneidenkäyttökykyyn ei ole tutkittu.
Cofact sisältää natriumia
Cofact sisältää enintään 448 mg natriumia (ruokasuolan toinen ainesosa) per 100 ml. Tämä vastaa 22 %:a suositellusta natriumin enimmäisvuorokausiannoksesta aikuiselle. Huomioi tämä, jos noudatat vähänatriumista ruokavaliota.
Miten valmistetta käytetään
Hoidon aloittaa, antaa ja sitä seuraa lääkäri, jolla on kokemusta hyytymishäiriöiden hoidosta. Lääkäri määrittää tarvittavan Cofact-annoksen verenohennuslääkkeiden käytöstä aiheutuneiden verenvuotojen hoitoon tai ennalta ehkäisyyn tai synnynnäisen K-vitamiinista riippuvien hyytymistekijöiden puutoksen hoitoon.
Annoksen suuruuteen vaikuttavat
- tilasi vakavuus
- painosi
- ne hyytymistekijät, joita tarvitset
- näiden tekijöiden pitoisuudet veressäsi.
Hyytymistekijöiden synnynnäisessä puutostilassa on tärkeää, että hyytymistekijöiden pitoisuudet veressä mitataan säännöllisesti.
Pakkausselosteen lopussa on tietoja terveydenhuollon ammattilaisille.
Jos käytät enemmän Cofact-valmistetta kuin sinun pitäisi
Lääkärin pitää tarkistaa veresi hyytymistilanne säännöllisesti hoidon aikana. Suuriin protrombiinikompleksikonsentraattiannoksiin on liittynyt sydäninfarkteja, disseminoitunutta intravaskulaarista koagulaatiota ja lisääntynyttä veritulppien muodostumista potilailla, joilla on tällaisten komplikaatioiden riski.
Mahdolliset haittavaikutukset
Kuten kaikki lääkkeet, tämäkin lääke voi aiheuttaa haittavaikutuksia. Kaikki eivät kuitenkaan niitä saa.
Seuraavia haittavaikutuksia on havaittu:
Yleinen (saattaa ilmaantua harvemmalle kuin 1 potilaalle kymmenestä):
- veritulppien muodostumisen riski (ks. kohta Mitä sinun on tiedettävä ennen valmisteen käyttöä).
Melko harvinainen (saattaa ilmaantua harvemmalle kuin 1 potilaalle sadasta):
- verenpaineen laskun riski.
Seuraavien haittavaikutusten esiintyvyys on tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin):
- yliherkkyys tai allergiset reaktiot (ks. kohta Mitä sinun on tiedettävä ennen valmisteen käyttöä)
- sydäninfarkti
- pahoinvointi, oksentelu
- infuusiokohdan punoitus, infuusiokohdan ärsytys, infuusiokohdan turpoaminen, huonovointisuus
- maksan toimintakoetulosten tilapäinen nousu
- aivohalvaus, heitehuimaus
- keuhkoembolia, hengitysvaikeus
- voimakas hikoilu, ihon kutina, nokkosihottuma, ihottuma.
Potilailla, joilla esiintyy hyytymistekijän II, VII, IX tai X puutostila, voi kehittyä Cofactin käytön seurauksena kyseisen tekijän vasta-aineita. Tällöin valmiste ei vaikuta toivotulla tavalla.
Haittavaikutuksista ilmoittaminen
Jos havaitset haittavaikutuksia, kerro niistä lääkärille tai apteekkihenkilökunnalle. Tämä koskee myös sellaisia mahdollisia haittavaikutuksia, joita ei ole mainittu tässä pakkausselosteessa. Voit ilmoittaa haittavaikutuksista myös suoraan (ks. yhteystiedot alla). Ilmoittamalla haittavaikutuksista voit auttaa saamaan enemmän tietoa tämän lääkevalmisteen turvallisuudesta.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Valmisteen säilyttäminen
Ei lasten ulottuville eikä näkyville.
Älä käytä tätä lääkettä pakkauksessa mainitun viimeisen käyttöpäivämäärän (EXP) jälkeen. Viimeinen käyttöpäivämäärä tarkoittaa kuukauden viimeistä päivää.
Säilytä jääkaapissa (2°C - 8°C). Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Cofactia voidaan säilyttää enintään 25 °C:ssa enintään kuuden kuukauden ajan esimerkiksi matkustamisen aikana eikä se vaikuta valmisteen tehoon. Päivämäärä, jolloin valmiste on otettu huoneenlämpöön, pitää merkitä pakkaukseen. Jos valmistetta ei käytetä kuuden kuukauden kuluessa siitä, kun se on otettu huoneenlämpöön, valmiste pitää hävittää.
Jääkaapista otettua valmistetta ei saa enää palauttaa jääkaappiin.
Liuotetun valmisteen säilyvyydeksi on osoitettu enintään 3 tuntia 15–25 ºC:ssa. Liuotettu valmiste pitää kontaminoitumisen estämiseksi kuitenkin käyttää heti.
Lääkkeitä ei tule heittää viemäriin eikä hävittää talousjätteiden mukana. Kysy käyttämättömien lääkkeiden hävittämisestä apteekista. Näin menetellen suojelet luontoa.
Pakkauksen sisältö ja muuta tietoa
Mitä Cofact sisältää
- Vaikuttavat aineet ovat hyytymistekijät II, VII, IX ja X ja muut vaikuttavat aineet ovat proteiini C ja proteiini S.
- Yksi Cofact 250 IU injektiopullo sisältää 250 IU tekijää IX; 140 - 350 IU tekijää II; 70 - 200 IU tekijää VII ja 140 - 350 IU tekijää X; 111–390 IU proteiinia C; 10–80 IU proteiinia S.
- Yksi Cofact 500 IU injektiopullo sisältää 500 IU tekijää IX; 280 - 700 IU tekijää II; 140 - 400 IU tekijää VII ja 280 - 700 IU tekijää X; 222–780 IU proteiinia C; 20–160 IU proteiinia S.
Kun kuiva-aine on sekoitettu pakkauksen sisältämään liuottimeen, käyttövalmis injektioneste sisältää:
- 14 - 35 IU/ml tekijää II
- 7 - 20 IU/ml tekijää VII
- 25 IU/ml tekijää IX
- 14 - 35 IU/ml tekijää X
- 11–39 IU/ml proteiinia C
- 1–8 IU/ml proteiinia S.
- Muut aineet ovat natriumsitraatti, natriumkloridi ja antitrombiini.
Lääkevalmisteen kuvaus ja pakkauskoot
Cofact injektiokuiva-aine on sinertävä jauhe. Käyttövalmis injektioneste on sinertävä liuos.
Cofact on injektiokuiva-aine ja liuotin, liuosta varten.
250 IU:n pakkauksen sisältö
yksi 250 IU kuiva-ainetta sisältävä injektiopullo
yksi 10 ml injektionesteisiin käytettävää vettä sisältävä injektiopullo
yksi siirtokanyyli.
500 IU:n pakkauksen sisältö
yksi 500 IU kuiva-ainetta sisältävä injektiopullo
yksi 20 ml injektionesteisiin käytettävää vettä sisältävä injektiopullo
yksi siirtokanyyli.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Myyntiluvan haltija ja valmistaja
Prothya Biosolutions Netherlands B.V.
Plesmanlaan 125
NL-1066 CX Amsterdam
Alankomaat
Tällä lääkkeellä on myyntilupa Euroopan talousalueeseen kuuluvissa jäsenvaltioissa seuraavilla kauppanimillä:
Itävalta, Belgia, Suomi, Ranska, Saksa, Islanti, Italia, Luxemburg, Alankomaat ja Espanja: Cofact.
Ruotsi: Thyaplex
Tämä pakkausseloste on tarkistettu viimeksi 22.11.2022
Ohjeet terveydenhuollon ammattilaiselle
Seuraavat tiedot on tarkoitettu vain terveydenhuollon ammattilaisille:
Vaikuttavat aineet ja niiden määrät
Cofact (neljän hyytymistekijän konsentraatti) on injektiokuiva-aine ja liuotin, liuosta varten, joka sisältää ihmisen protrombiinikompleksia. Valmiste sisältää nimellisesti seuraavassa taulukossa esitetyt määrät ihmisen hyytymistekijöitä:
|
|
Cofact 250 IU (tekijä IX) |
Cofact 500 IU (tekijä IX) |
Käyttövalmiiksi liuottamisen jälkeen* (IU/ml) |
|
Vaikuttavat aineet |
|
|
|
|
Hyytymistekijä II |
140–350 |
280–700 |
14–35 |
|
Hyytymistekijä VII |
70–200 |
140–400 |
7–20 |
|
Hyytymistekijä IX |
250 |
500 |
25 |
|
Hyytymistekijä X |
140–350 |
280–700 |
14–35 |
|
Muut vaikuttavat aineet |
111–390 10–80 |
222–780 20–160 |
11–39 1–8 |
|
Proteiini C Proteiini S |
|||
|
*10 ml:lla (Cofact 250 IU) tai 20 ml:lla (Cofact 500 IU) injektionesteisiin käytettävää vettä käyttökuntoon saatettuna |
|||
Kokonaisproteiinimäärä injektiopulloa kohti on 130–350 mg (Cofact 250 IU) tai 260–700 mg (Cofact 500 IU). Valmisteen spesifinen aktiivisuus on ≥ 0,6 IU/mg tekijä IX ‑aktiivisuutena ilmaistuna.
Kaikkien hyytymistekijöiden sekä proteiini C:n ja S:n (antigeeni) aktiivisuus on testattu WHO:n tai Euroopan farmakopean voimassa olevien standardien mukaisesti.
Käyttökuntoon saatettuna tämä lääkevalmiste sisältää 125–195 mmol/l natriumia, enintään 44,8 mg natriumia per 10 ml.
Annostus ja antotapa
Annostus
Seuraavassa on esitetty ainoastaan yleiset annostusohjeet. Hoito tulee aloittaa hyytymishäiriöiden hoitoon erikoistuneen lääkärin valvonnassa. Annostus ja korvaushoidon kesto riippuvat häiriön vakavuudesta, vuodon sijainnista ja laajuudesta sekä potilaan kliinisestä tilasta.
Annettava määrä ja antotiheys on laskettava kunkin potilaan kohdalla yksilöllisesti. Annosvälit tulee sovittaa protrombiinikompleksiin kuuluvien hyytymistekijöiden eripituisten verenkierron puoliintumisaikojen mukaisesti. Yksilölliset annostarpeet on mahdollista tunnistaa ainoastaan määrittämällä kyseessä olevien hyytymistekijöiden plasmapitoisuudet säännöllisesti tai määrittämällä protrombiinikompleksipitoisuudet (protrombiiniaika, INR) yleistestien avulla ja seuraamalla tiiviisti potilaan kliinistä tilaa.
Suurissa kirurgisissa toimenpiteissä on välttämätöntä seurata korvaushoitoa tarkkaan hyytymistekijäpitoisuusmääritysten avulla (spesifiset hyytymistekijäpitoisuusmääritykset ja/tai protrombiinikompleksipitoisuuksien yleistestit).
Verenvuodot ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy K-vitamiiniantagonistihoidon aikana:
Annos riippuu hoitoa edeltävästä INR-arvosta, tavoitteena olevasta INR-arvosta ja potilaan painosta. Seuraavissa taulukoissa on annettu suuntaa-antavat annokset INR-arvon korjaamiseksi INR:n eri lähtöarvoilla.
Annostaulukot edustavat ainoastaan yleisiä annostusohjeita. Ne eivät korvaa yksilöllisen annoksen arviointia kunkin potilaan kohdalla eivätkä hoidon aikaista INR-arvon ja muiden hyytymisparametrien huolellista seurantaa.
Cofactin annossuositukset (ml) tavoite-INR-arvon ≤ 2,1 saavuttamiseksi
| Lähtö-INR | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Potilaan paino | ||||||||||||
| 50 kg | 40 | 40 | 40 | 30 | 30 | 30 | 20 | 20 | x | x | x | x |
| 60 kg | 50 |
50 |
40 | 40 | 30 | 30 | 30 | 20 | x | x | x | x |
| 70 kg | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | x | x | x | x |
| 80 kg | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 30 | x | x | x | x |
| 90 kg | 60 | 60 | 60 | 60 | 50 | 50 | 40 | 30 | x | x | x | x |
| 100 kg | 60 | 60 | 60 | 60 | 60 | 50 | 40 | 40 | x | x | x | x |
Cofactin annossuositukset (ml) tavoite-INR-arvon ≤ 1,5 saavuttamiseksi
| Lähtö-INR | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Potilaan paino | ||||||||||||
| 50 kg | 60 | 60 | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 30 |
| 60 kg | 80 |
70 |
70 | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 40 | 30 |
| 70 kg | 90 | 80 | 80 | 70 | 70 | 70 | 60 | 60 | 50 | 40 | 40 | 40 |
| 80 kg | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 50 | 40 |
| 90 kg | 100 | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 40 |
| 100 kg | 100 | 100 | 100 | 100 | 100 | 90 | 90 | 80 | 70 | 70 | 60 | 50 |
Annokset on laskettu Cofactin tekijä IX ‑pitoisuuden perusteella, koska tällä on suhteellisen lyhyt puoliintumisaika ja alhainen infuusion jälkeinen saanto verrattuna muiden protrombiinikompleksin hyytymistekijöiden vastaaviin ominaisuuksiin. Oletuksena on, että tekijä IX:n keskimääräinen plasmapitoisuus ≥ 30 % on riittävä INR-arvon ≤ 2,1 saavuttamiseksi ja ≥ 60 % on riittävä INR-arvon ≤ 1,5 saavuttamiseksi. Laskennalliset määrät on pyöristetty alaspäin 10 ml:n kerrannaisiksi, ja ylärajaksi on asetettu 60 ml tai 100 ml (katso edellä olevat taulukot). Tavoite-INR-arvot ovat Federation of Dutch Thrombosis Services -organisaation suosittelemat ja samankaltaiset kuin englantilaiset ja saksalaiset suositukset.
K-vitamiiniantagonistin aiheuttaman heikentyneen hemostaasin korjaaminen kestää noin 6–8 tuntia. Samaan aikaan annetun K-vitamiinin vaikutukset saavutetaan yleensä kuitenkin 4–6 tunnin kuluessa. Näin ollen ihmisen protrombiinikompleksia ei yleensä tarvitse antaa toistuvasti silloin, kun annetaan K-vitamiinia.
Koska mainitut suositukset ovat kokemusperäisiä ja potilaan tilan paraneminen ja vaikutuksen kesto saattavat vaihdella, INR-arvoa on seurattava hoidon aikana.
Verenvuodot ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy synnynnäisessä, jonkin K-vitamiinista riippuvan hyytymistekijän puutoksessa, kun spesifistä hyytymistekijää ei ole saatavilla:
Tarvittava laskennallinen hoitoannos perustuu kokemusperäiseen havaintoon. Sen mukaan noin 1 IU tekijää VII tai tekijää IX potilaan painokiloa kohti nostaa plasman tekijä VII ‑aktiivisuutta tai tekijä IX -aktiivisuutta vastaavasti 0,01 IU/ml. Edelleen 1 IU tekijää II tai tekijää X potilaan painokiloa kohti nostaa plasman tekijä II -aktiivisuutta 0,02 IU/ml tai tekijä X -aktiivisuutta 0,017 IU/ml.
Kunkin hyytymistekijän annos ilmaistaan kansainvälisinä yksikköinä (International Unit, IU), jotka perustuvat kyseisen tekijän voimassaolevaan WHO-standardiin. Yksittäisen hyytymistekijän aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliplasmaan) tai kansainvälisinä yksikköinä (suhteessa kyseisen hyytymistekijän kansainväliseen standardiin).
Yksi kansainvälinen yksikkö (IU) hyytymistekijäaktiivisuutta vastaa kyseisen tekijän määrää 1 ml:ssa normaalia ihmisplasmaa.
Esimerkiksi tarvittavan tekijä X:n annoksen laskeminen perustuu kokemusperäiseen havaintoon, jonka mukaan 1 IU tekijää X potilaan painokiloa kohti nostaa plasman tekijä X ‑aktiivisuutta 0,017 IU/ml. Vaadittava annos määritetään seuraavan kaavan avulla:
Tarvittava yksikkömäärä = potilaan paino (kg) x haluttu tekijä X ‑pitoisuuden nousu (IU/ml) x 60
jossa 60 (ml/kg) on arvioidun saannon käänteisluku.
Jos yksilöllinen saanto on tiedossa, laskennassa pitää käyttää kyseistä arvoa.
Antotapa
Ks. kohdasta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa. Cofact annetaan laskimoon.
Käyttövalmiin tuotteen suositeltu antonopeus on noin 2 ml/min.
Pediatriset potilaat
Cofact-valmisteen käytön turvallisuutta ja tehoa pediatrisille potilaille ei ole varmistettu.
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Cofact on yhteensopiva polypropyleenimateriaalin kanssa. Hyytymistekijän adsorptio muiden injektio- ja infuusiovälineiden sisäpintaan saattaa aiheuttaa hoidon epäonnistumisen.
Kestoaika
3 vuotta.
Käyttökuntoon saatetun valmisteen kemialliseksi ja fysikaaliseksi käytönaikaiseksi säilyvyydeksi on osoitettu 3 tuntia 15–25 °C:n lämpötilassa. Mikrobiologiselta kannalta valmiste pitää käyttää heti käyttökuntoon saattamisen jälkeen. Jos sitä ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet
Liuottaminen
Kuivattu proteiinifraktio tulee liuottaa annettuun tilavuuteen injektionesteisiin käytettävää vettä. Jos Cofact-injektiopullo ja liuotinpullo on säilytetty 2 °C – 8 °C:ssa, ne on hyvä saattaa huoneenlämpöisiksi (15 °C – 25 °C) ennen liuottamista.
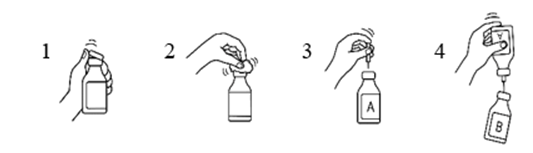
Siirtokanyylin käyttö
- Poista vesipullon ja kuiva-ainepullon muovisuojukset.
- Desinfioi pullojen kumitulpat 70‑prosenttiseen alkoholiin kostutetulla sideharsolapulla.
- Poista siirtokanyylin toisen pään suojus. Työnnä neula vesipulloon (A).
- Poista sitten siirtokanyylin toisen pään suojus, käännä siirtokanyylissa kiinni oleva pullo ylösalaisin ja työnnä välittömästi vapaana oleva neula kuiva-ainepulloon (B).
Kuiva-ainepullossa oleva alipaine imee veden pulloon. Suositus: veden valuessa kuiva-ainepulloa tulee pitää kallistettuna, jolloin vesi valuu pullon seinää pitkin. Tämä nopeuttaa tuotteen liukenemista. Heti kun koko vesimäärä on valunut pulloon, tyhjä pullo ja siirtokanyyli tulee poistaa samanaikaisesti.
Liukenemistapahtumaa voidaan nopeuttaa pyörittämällä pulloa varovasti ja, mikäli tarpeen, lämmittämällä se 30 °C:een. Pulloa ei koskaan saa ravistaa eikä lämmittää yli 37 °C:seen. Jos pulloa lämmitetään vesihauteessa, tulee varmistaa, että vesi ei pääse kosketuksiin suojuksen ja/tai kumitulpan kanssa.
Pääsääntöisesti kuiva-aineen tulisi olla täysin liuennut 10 minuutin kuluessa muodostaen sinisen liuoksen; sininen väri johtuu plasmaproteiini keruloplasmiinista.
Liuoksen tulee olla kirkas tai hieman opalisoiva. Älä käytä sameaa tai sakkaista liuosta. Käyttövalmiiksi liuotettu tuote tulee tarkastaa visuaalisesti hiukkasten ja värimuutosten osalta ennen antamista.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.